Having cancer brings many changes to a child’s life. You can help your child by keeping her life as normal as possible.
Credit: iStock
A cancer diagnosis is upsetting at any age, but especially so when the patient is a child. It’s natural to have many questions, such as, Who should treat my child? Will my child get well? What does all of this mean for our family? Not all questions have answers, but the information and resources on this page provide a starting point for understanding the basics of childhood cancer.
Types of Cancer in Children
In the United States in 2024, an estimated 9,620 new cases of cancer will be diagnosed among children from birth to 14 years, and about 1,040 children are expected to die from the disease. Although cancer death rates for this age group have declined by 70 percent from 1970 through 2020, cancer remains the leading cause of death from disease among children. The most common types of cancer diagnosed in children ages 0 to 14 years are leukemias, brain and other central nervous system (CNS) tumors, and lymphomas.
NCI’s Cancer Stat Facts include detailed cancer rate and trend information for certain types of childhood cancer.
Treating Childhood Cancer
Children’s cancers are not always treated like adult cancers. Pediatric oncology is a medical specialty focused on the care of children with cancer. It’s important to know that this expertise exists and that there are effective treatments for many childhood cancers.
Treatment Considerations for Children with Cancer
Pediatric oncology experts and parents discuss childhood cancer treatment-related decisions, side effects, clinical trials for children with cancer, and strategies to care for children at home.
There are many types of cancer treatment. The types of treatment that a child with cancer receives will depend on the type of cancer and how advanced it is. Common treatments include: surgery, chemotherapy, radiation therapy, immunotherapy, and stem cell transplant. Learn about these and other therapies in our Types of Treatment section.
Our summary about Childhood Cancer Genomics describes the genomic alterations associated with different pediatric cancers, and their significance for therapy and prognosis.
Before any new treatment can be made widely available to patients, it must be studied in clinical trials (research studies) and found to be safe and effective in treating disease. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials.
Children face unique issues during their treatment for cancer, after the completion of treatment, and as survivors of cancer. For example, they may receive more intense treatments, cancer and its treatments have different effects on growing bodies than adult bodies, and they may respond differently to drugs that control symptoms in adults. Late effects of treatment are discussed later on this page in the Survivorship section.
Where Children with Cancer Are Treated
Children who have cancer are often treated at a children’s cancer center, which is a hospital or unit in a hospital that specializes in treating children with cancer.
The doctors and other health professionals at these centers have special training and expertise to give complete care to children. Specialists at a children’s cancer center are likely to include primary care physicians, pediatric oncologists/hematologists, pediatric surgical specialists, radiation oncologists, rehabilitation specialists, pediatric nurse specialists, social workers, and psychologists. At these centers, clinical trials are available for most types of cancer that occur in children, and the opportunity to participate in a trial is offered to many patients.
Hospitals that have experts in treating children with cancer are usually member institutions of the NCI-supported Children’s Oncology Group (COG). COG is the world’s largest organization that conducts clinical research to improve the care and treatment of children with cancer. NCI’s Cancer Information Service can help families find COG-affiliated hospitals.
At the NIH Clinical Center in Bethesda, Maryland, NCI’s Pediatric Oncology Branch cares for children and young adults with cancer. Health professionals and scientists conduct translational research that spans basic science to clinical trials to improve outcomes for children and young adults with cancer and genetic tumor predisposition syndromes.
Coping with Cancer
Adjusting to a child’s cancer diagnosis and finding ways to stay strong is challenging for everyone in a family. Our page, Support for Families When a Child Has Cancer, has tips for talking with children about their cancer and preparing them for changes they may experience. Also included are ways to help brothers and sisters cope, steps parents can take when they need support, and tips for working with the health care team. Various aspects of coping and support are also discussed in the publication Children with Cancer: A Guide for Parents.
Survivorship
It’s essential for childhood cancer survivors to receive follow-up care to monitor their health after completing treatment. All survivors should have a treatment summary and a survivorship care plan, as discussed on our Care for Childhood Cancer Survivors page. That page also has information on clinics that specialize in providing follow-up care for people who have had childhood cancer.
Survivors of any kind of cancer can develop health problems months or years after cancer treatment, known as late effects, but late effects are of particular concern for childhood cancer survivors because treatment of children can lead to profound, lasting physical and emotional effects. Late effects vary with the type of cancer, the child’s age, the type of treatment, and other factors. Information on types of late effects and ways to manage these can be found on our Care for Childhood Cancer Survivors page. The PDQ® Late Effects of Treatment for Childhood Cancer summary has in-depth information.
The causes of most childhood cancers are not known. About 8 to 10 percent of all cancers in children are caused by an inherited mutation (a genetic mutation that can be passed from parents to their children).
Most cancers in children, like those in adults, are thought to develop as a result of mutations in genes that lead to uncontrolled cell growth and eventually cancer. In adults, these gene mutations reflect the cumulative effects of aging and long-term exposure to cancer-causing substances. However, identifying potential environmental causes of childhood cancer has been difficult, partly because cancer in children is rare and partly because it is difficult to determine what children might have been exposed to early in their development. More information about possible causes of cancer in children is available in the fact sheet, Cancer in Children and Adolescents.
Research
NCI supports a broad range of research to better understand the causes, biology, and patterns of childhood cancers and to identify the best ways to successfully treat children with cancer. In the context of clinical trials, researchers are treating and learning from young cancer patients. Researchers are also following childhood cancer survivors to learn about health and other issues they may face as a result of their cancer treatment. To learn more, see Childhood Cancers Research.
Targeted therapy treats cancer by targeting proteins that control how cancer cells grow, divide, and spread.
Credit: National Cancer Institute
What is targeted therapy?
Targeted therapy is a type of cancer treatment that targets proteins that control how cancer cells grow, divide, and spread. It is the foundation of precision medicine. As researchers learn more about the DNA changes and proteins that drive cancer, they are better able to design treatments that target these proteins.
Small-molecule drugs are small enough to enter cells easily, so they are used for targets that are inside cells.
Monoclonal antibodies, also known as therapeutic antibodies, are proteins produced in the lab. These proteins are designed to attach to specific targets found on cancer cells. Some monoclonal antibodies mark cancer cells so that they will be better seen and destroyed by the immune system. Other monoclonal antibodies directly stop cancer cells from growing or cause them to self-destruct. Still others carry toxins to cancer cells. Learn more about monoclonal antibodies.
How Monoclonal Antibodies Treat Cancer
Learn how monoclonal antibodies such as trastuzumab, pembrolizumab, and rituximab are used to treat cancer.
Who is treated with targeted therapy?
For some types of cancer, such as chronic myelogenous leukemia (also known as CML), most people with that cancer will have a target for a certain drug, so they can be treated with that drug. But most of the time, your tumor will need to be tested to see if it contains targets for which there is a drug.
Testing your cancer for targets that could help choose your treatment is called biomarker testing. See Biomarker Testing for Cancer Treatment for more information.
You may need to have a biopsy for biomarker testing. A biopsy is a procedure in which your doctor removes a piece of the tumor for testing. There are some risks to having a biopsy. These risks vary depending on the size of the tumor and where it is located. Your doctor will explain the risks of having a biopsy for your type of tumor.
Most types of targeted therapy help treat cancer by interfering with specific proteins that help tumors grow and spread throughout the body. This is different from chemotherapy, which often kills all cells that grow and divide quickly. The following explains the different ways that targeted therapy treats cancer.
Help the immune system destroy cancer cells. One reason that cancer cells thrive is because they can hide from your immune system. Certain targeted therapies can mark cancer cells so it is easier for the immune system to find and destroy them. Other targeted therapies help boost your immune system to work better against cancer. Learn more about immunotherapy to treat cancer.
Stop cancer cells from growing by interrupting signals that cause them to grow and divide without order. Healthy cells in your body usually divide to make new cells only when they receive strong signals to do so. These signals bind to proteins on the cell surface, telling the cells to divide. This process helps new cells form only as your body needs them. But, some cancer cells have changes in the proteins on their surface that tell them to divide whether or not signals are present. Some targeted therapies interfere with these proteins, preventing them from telling the cells to divide. This process helps slow cancer’s uncontrolled growth.
Stop signals that help form blood vessels. To grow beyond a certain size, tumors need to form new blood vessels in a process called angiogenesis. The tumor sends signals that start angiogenesis. Some targeted therapies called angiogenesis inhibitors interfere with these signals to prevent a blood supply from forming. Without a blood supply, tumors stay small. Or, if a tumor already has a blood supply, these treatments can cause blood vessels to die, which causes the tumor to shrink. Learn more about angiogenesis inhibitors.
Deliver cell-killing substances to cancer cells. Some monoclonal antibodies are combined with cell-killing substances such as toxins, chemotherapy drugs, or radiation. Once these monoclonal antibodies attach to targets on the surface of cancer cells, the cells take up the cell-killing substances, causing them to die. Cells that don’t have the target will not be harmed.
Cause cancer cell death. Healthy cells die in an orderly manner when they become damaged or are no longer needed. But, cancer cells have ways of avoiding this dying process. Some targeted therapies can cause cancer cells to go through this process of cell death, which is called apoptosis.
Starve cancer of hormones it needs to grow. Some breast and prostate cancers require certain hormones to grow. Hormone therapies are a type of targeted therapy that can work in two ways. Some hormone therapies prevent your body from making specific hormones. Others prevent the hormones from acting on your cells, including cancer cells. Learn more about hormone therapy for prostate cancer and hormone therapy for breast cancer.
Are there drawbacks to targeted therapy?
Targeted therapy does have some drawbacks.
Cancer cells can become resistant to targeted therapy. Resistance can happen when the target itself changes and the targeted therapy is not able to interact with it. Or it can happen when cancer cells find new ways to grow that do not depend on the target. Because of resistance, targeted therapy may work best when used with more than one type of targeted therapy or with other cancer treatments, such as chemotherapy and radiation.
Drugs for some targets are hard to develop. Reasons include the target’s structure, the target’s function in the cell, or both.
What are the side effects of targeted therapy?
When targeted therapy was first developed, scientists thought that it would be less toxic than chemotherapy. But they have learned that targeted therapy can also cause serious side effects. The side effects that you may have depends on the type of targeted therapy you receive and how your body reacts to it.
The most common side effects of targeted therapy include diarrhea and liver problems. Other side effects might include
Small-molecule drugs are pills or capsules that you can swallow.
Monoclonal antibodies are usually given through a needle in a blood vein.
Where do I go for targeted therapy?
Where you go for treatment depends on which drugs you are getting and how they are given. You may take targeted therapy at home. Or you may receive targeted therapy in a doctor’s office, clinic, or outpatient unit in a hospital. Outpatient means you do not spend the night in the hospital.
How often will I receive targeted therapy?
How often and how long you receive targeted therapy depends on
your type of cancer and how advanced it is
the type of targeted therapy
how your body reacts to treatment
You may have treatment every day, every week, or every month. Some targeted therapies are given in cycles. A cycle is a period of treatment followed by a period of rest. The rest period gives your body a chance to recover and build new healthy cells.
How will targeted therapy affect me?
Targeted therapy affects people in different ways. How you feel depends on how healthy you are before treatment, your type of cancer, how advanced it is, the kind of targeted therapy you are getting, and the dose. Doctors and nurses cannot know for certain how you will feel during treatment.
How will I know whether targeted therapy is working?
While you are receiving targeted therapy, you will see your doctor often. He or she will give you physical exams and ask you how you feel. You will have medical tests, such as blood tests, x-rays, and different types of scans. These regular visits and tests will help the doctor know whether the treatment is working.
Where can I find out about clinical trials of targeted therapy?
Clinical trials of targeted therapy and other cancer treatments take place in cities and towns across the United States and throughout the world. They take place in doctors’ offices, cancer centers, medical centers, community hospitals and clinics, and veteran and military hospitals.
To find clinical trials of targeted therapy use this advanced search form. Under “Keywords/Phrases,” type “targeted therapy.” Under “Trial Type,” select the box for “Treatment” trials.
Carcinoma of unknown primary is a disease in which malignant (cancer) cells are found in the body but the place the cancer began is not known.
The signs and symptoms of disease are caused by the metastatic cancer and depend on where the cancer has spread.
Blood and imaging tests are done to learn more about the cause of the signs and symptoms.
A biopsy is done to diagnose metastatic cancer.
Because the place where the cancer started is not known, more tests and procedures are done to search for the primary cancer.
When tests are able to find the primary cancer, the cancer is no longer a CUP and treatment is based on the type of primary cancer.
Sometimes the primary cancer is never found.
Carcinoma of unknown primary is a disease in which malignant (cancer) cells are found in the body but the place the cancer began is not known.
Cancer can form in any tissue in the body. The first cancer to form is called the primary cancer. The process of cancer cells spreading to other parts of the body is called metastasis. The cancer that has spread to another part of the body is called metastatic cancer.
EnlargeIn cancer of unknown primary, cancer cells have spread in the body but the place where the primary cancer began is not known.
Many cancer deaths are caused when cancer moves from the original tumor and spreads to other tissues and organs. This is called metastatic cancer. This animation shows how cancer cells travel from the place in the body where they first formed to other parts of the body.
The signs and symptoms of disease are caused by the metastatic cancer and depend on where the cancer has spread.
The primary cancer does not cause signs and symptoms of disease.
Check with your doctor if you have any of the following general signs of cancer:
Lump or thickening in any part of the body.
Pain that is in one part of the body and does not go away.
A cough that does not go away or hoarseness in the voice.
The pathologist views the tissue to look for cancer cells and to find out the type of cancer. Cancer cells usually look like the cells in the type of tissue in which the cancer began. In CUP, the cancer cells do not look like the cells of the tissue they were found in. The pathologist cannot determine the type of primary cancer.
One or more of the following laboratory tests may be used to further study the tissue samples:
Genetic analysis: A laboratory test in which the DNA in a sample of cancer cells or tissue is studied to check for mutations (changes) that may help predict the best treatment for CUP.
Histologic study: A laboratory test in which stains are added to a sample of cancer cells or tissue and viewed under a microscope to look for certain changes in the cells. Certain changes in the cells are linked to certain types of cancer.
Immunohistochemistry: A laboratory test that uses antibodies to check for certain antigens (markers) in a sample of a patient’s tissue. The antibodies are usually linked to an enzyme or a fluorescent dye. After the antibodies bind to a specific antigen in the tissue sample, the enzyme or dye is activated, and the antigen can then be seen under a microscope. This type of test is used to help diagnose cancer and to help tell one type of cancer from another type of cancer.
Reverse transcription–polymerase chain reaction (RT–PCR) test: A laboratory test in which the amount of a genetic substance called mRNA made by a specific gene is measured. An enzyme called reverse transcriptase is used to convert a specific piece of RNA into a matching piece of DNA, which can be amplified (made in large numbers) by another enzyme called DNA polymerase. The amplified DNA copies help tell whether a specific mRNA is being made by a gene. RT–PCR can be used to check the activation of certain genes that may indicate the presence of cancer cells. This test may be used to look for certain changes in a gene or chromosome, which may help diagnose cancer.
Cytogenetic analysis: A laboratory test in which the chromosomes of cells in a sample of tumor tissue are counted and checked for any changes, such as broken, missing, rearranged, or extra chromosomes. Changes in certain chromosomes may be a sign of cancer. Cytogenetic analysis is used to help diagnose cancer, plan treatment, or find out how well treatment is working. Changes in certain chromosomes are linked to certain types of cancer.
Light and electron microscopy: A laboratory test in which cells in a sample of tissue are viewed under regular and high-powered microscopes to look for certain changes in the cells.
Because the place where the cancer started is not known, more tests and procedures are done to search for the primary cancer.
The following tests and procedures may be done:
Physical exam and health history: An exam of the body to check general signs of health, including checking for signs of disease, such as lumps or anything else that seems unusual. A history of the patient’s health habits and past illnesses and treatments will also be taken.
CT scan (CAT scan): A procedure that makes a series of detailed pictures of areas inside the body, such as the chest or abdomen, taken from different angles. The pictures are made by a computer linked to an x-ray machine. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography.
MRI (magnetic resonance imaging): A procedure that uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the body. This procedure is also called nuclear magnetic resonance imaging (NMRI).
PET scan (positron emission tomography scan): A procedure to find malignant tumor cells in the body. A small amount of radioactive glucose (sugar) is injected into a vein. The PET scanner rotates around the body and makes a picture of where glucose is being used in the body. Malignant tumor cells show up brighter in the picture because they are more active and take up more glucose than normal cells do.
Ultrasound exam: A procedure in which high-energy sound waves (ultrasound) are bounced off internal tissues or organs and make echoes. The echoes form a picture of body tissues called a sonogram. The picture can be printed to be looked at later.
Endoscopy: A procedure to look at organs and tissues inside the body to check for abnormal areas. An endoscope is inserted through an incision (cut) in the skin or opening in the body, such as the mouth. An endoscope is a thin, tube-like instrument with a light and a lens for viewing. It may also have a tool to remove tissue or lymph node samples, which are checked under a microscope for signs of disease. For example, a colonoscopy may be done.
The primary cancer was removed during surgery for another condition and doctors didn’t know cancer had formed. For example, in adults, a uterus with cancer may be removed during a hysterectomy to treat a serious infection.
Because the primary cancer is unknown, it may be harder to choose the best treatment.
Stages of Childhood Carcinoma of Unknown Primary
The extent or spread of cancer is usually described as stages. The stage of the cancer is usually used to plan treatment. However, carcinoma of unknown primary (CUP) has already spread to other parts of the body when it is found. There is no standard staging system for CUP.
Sometimes childhood carcinoma of unknown primary recurs (comes back) after treatment.
Treatment Option Overview
Key Points
There are different types of treatment for children with carcinoma of unknown primary.
Children with carcinoma of unknown primary should have their treatment planned by a team of doctors who are experts in treating childhood cancer.
Three types of standard treatment are used:
Radiation therapy
Chemotherapy
Targeted therapy
New types of treatment are being tested in clinical trials.
Treatment for childhood carcinoma of unknown primary may cause side effects.
Patients may want to think about taking part in a clinical trial.
Patients can enter clinical trials before, during, or after starting their cancer treatment.
Follow-up tests may be needed.
There are different types of treatment for children with carcinoma of unknown primary.
Some treatments are standard (the currently used treatment), and some are being tested in clinical trials. A treatment clinical trial is a research study meant to help improve current treatments or obtain information on new treatments for patients with cancer. When clinical trials show that a new treatment is better than the standard treatment, the new treatment may become the standard treatment.
Because cancer in children is rare, taking part in a clinical trial should be considered. Some clinical trials are open only to patients who have not started treatment.
Children with carcinoma of unknown primary should have their treatment planned by a team of doctors who are experts in treating childhood cancer.
Treatment will be overseen by a pediatric oncologist, a doctor who specializes in treating children with cancer. The pediatric oncologist works with other pediatric health professionals who are experts in treating children with cancer and who specialize in certain areas of medicine. This may include the following specialists and others:
Chemotherapy is a cancer treatment that uses drugs to stop the growth of cancer cells, either by killing the cells or by stopping them from dividing. When chemotherapy is taken by mouth or injected into a vein or muscle, the drugs enter the bloodstream and can reach cancer cells throughout the body (systemic chemotherapy).
Targeted therapy
Targeted therapy is a treatment that uses drugs or other substances to identify and attack cancer cells.Targeted therapies usually cause less harm to normal cells than chemotherapy and radiation therapy do.
New types of treatment are being tested in clinical trials.
Patients may want to think about taking part in a clinical trial.
For some patients, taking part in a clinical trial may be the best treatment choice. Clinical trials are part of the cancer research process. Clinical trials are done to find out if new cancer treatments are safe and effective or better than the standard treatment.
Many of today’s standard treatments for cancer are based on earlier clinical trials. Patients who take part in a clinical trial may receive the standard treatment or be among the first to receive a new treatment.
Patients who take part in clinical trials also help improve the way cancer will be treated in the future. Even when clinical trials do not lead to effective new treatments, they often answer important questions and help move research forward.
Patients can enter clinical trials before, during, or after starting their cancer treatment.
Some clinical trials only include patients who have not yet received treatment. Other trials test treatments for patients whose cancer has not gotten better. There are also clinical trials that test new ways to stop cancer from recurring (coming back) or reduce the side effects of cancer treatment.
Clinical trials are taking place in many parts of the country. Information about clinical trials supported by NCI can be found on NCI’s clinical trials search webpage. Clinical trials supported by other organizations can be found on the ClinicalTrials.gov website.
Follow-up tests may be needed.
As your child goes through treatment, they will have follow-up tests or check-ups. Some tests that were done to diagnose or stage the cancer may be repeated to see how well the treatment is working. Decisions about whether to continue, change, or stop treatment may be based on the results of these tests.
Some of the tests will continue to be done from time to time after treatment has ended. The results of these tests can show if your child’s condition has changed or if the cancer has recurred (come back).
Treatment of Childhood Carcinoma of Unknown Primary
Sometimes childhood carcinoma of unknown primary can recur (come back) after treatment. If your child is diagnosed with a recurrent carcinoma of unknown primary, your child’s doctor will work with you to plan treatment.
Use our clinical trial search to find NCI-supported cancer clinical trials that are accepting patients. You can search for trials based on the type of cancer, the age of the patient, and where the trials are being done. General information about clinical trials is also available.
To Learn More About Childhood Carcinoma of Unknown Primary
Physician Data Query (PDQ) is the National Cancer Institute’s (NCI’s) comprehensive cancer information database. The PDQ database contains summaries of the latest published information on cancer prevention, detection, genetics, treatment, supportive care, and complementary and alternative medicine. Most summaries come in two versions. The health professional versions have detailed information written in technical language. The patient versions are written in easy-to-understand, nontechnical language. Both versions have cancer information that is accurate and up to date and most versions are also available in Spanish.
PDQ is a service of the NCI. The NCI is part of the National Institutes of Health (NIH). NIH is the federal government’s center of biomedical research. The PDQ summaries are based on an independent review of the medical literature. They are not policy statements of the NCI or the NIH.
Purpose of This Summary
This PDQ cancer information summary has current information about the treatment of childhood carcinoma of unknown primary. It is meant to inform and help patients, families, and caregivers. It does not give formal guidelines or recommendations for making decisions about health care.
Reviewers and Updates
Editorial Boards write the PDQ cancer information summaries and keep them up to date. These Boards are made up of experts in cancer treatment and other specialties related to cancer. The summaries are reviewed regularly and changes are made when there is new information. The date on each summary (“Updated”) is the date of the most recent change.
The information in this patient summary was taken from the health professional version, which is reviewed regularly and updated as needed, by the PDQ Pediatric Treatment Editorial Board.
Clinical Trial Information
A clinical trial is a study to answer a scientific question, such as whether one treatment is better than another. Trials are based on past studies and what has been learned in the laboratory. Each trial answers certain scientific questions in order to find new and better ways to help cancer patients. During treatment clinical trials, information is collected about the effects of a new treatment and how well it works. If a clinical trial shows that a new treatment is better than one currently being used, the new treatment may become “standard.” Patients may want to think about taking part in a clinical trial. Some clinical trials are open only to patients who have not started treatment.
Clinical trials can be found online at NCI’s website. For more information, call the Cancer Information Service (CIS), NCI’s contact center, at 1-800-4-CANCER (1-800-422-6237).
Permission to Use This Summary
PDQ is a registered trademark. The content of PDQ documents can be used freely as text. It cannot be identified as an NCI PDQ cancer information summary unless the whole summary is shown and it is updated regularly. However, a user would be allowed to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks in the following way: [include excerpt from the summary].”
Images in this summary are used with permission of the author(s), artist, and/or publisher for use in the PDQ summaries only. If you want to use an image from a PDQ summary and you are not using the whole summary, you must get permission from the owner. It cannot be given by the National Cancer Institute. Information about using the images in this summary, along with many other images related to cancer can be found in Visuals Online. Visuals Online is a collection of more than 3,000 scientific images.
Disclaimer
The information in these summaries should not be used to make decisions about insurance reimbursement. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s E-mail Us.
Cancers of unknown primary sites present as metastatic cancers for which precise primary tumor sites cannot be determined.[1] As an example, lymph nodes at the base of the skull may enlarge in relationship to a tumor on the face or scalp that is not evident by physical examination or radiographic imaging. Thus, modern imaging techniques may indicate the extent of the disease but not a primary site. Tumors such as adenocarcinomas, melanomas, and embryonal tumors, like rhabdomyosarcomas and neuroblastomas, may present in this way.
Less than 1% of all solid cancers of unknown primary sites occur in children. Because of the age-related incidence of tumor types, embryonal histologies are more common in children.[2]
References
Kuttesch JF, Parham DM, Kaste SC, et al.: Embryonal malignancies of unknown primary origin in children. Cancer 75 (1): 115-21, 1995. [PUBMED Abstract]
Pavlidis N, Pentheroudakis G: Cancer of unknown primary site. Lancet 379 (9824): 1428-35, 2012. [PUBMED Abstract]
Diagnostic Evaluation
For all patients who present with tumors from unknown primary sites, treatment is directed toward the specific histopathology of the tumor and is age-appropriate for the general type of cancer suspected, regardless of the sites involved.[1]
Studies in adults suggest that positron emission tomography (PET) imaging can be helpful in identifying cancers of unknown primary sites, particularly in patients whose tumors arise in the head and neck area.[2] A report in adults using fluorine F 18-fludeoxyglucose PET-computed tomography identified 42.5% of primary tumors in a group of cancers of unknown primary sites.[3]
The use of gene expression profiling and next-generation sequencing can enhance the ability to identify the putative tissue of origin and guide the selection of targeted agents for specific variants.[4–8]
In a study of 200 adult patients with carcinomas of unknown primary sites, 125 had adenocarcinomas and 75 had carcinomas without features of adenocarcinoma. Genomic alterations were found in 96% of the cases. The most common alterations were TP53 (55%), KRAS (20%), CDKN2A (19%), and MYC (12%). Clinically relevant and potentially actionable variants included KRAS (20%), CDKN2A (19%), MCL1 (10%), PTEN (7%), PIK3CA (9%), ERBB2 (8%), RICTOR (6%), BRAF (6%), and NF1 (4%). These findings suggest that genomic profiling can help identify potentially actionable targets, which could benefit patients clinically while reducing the complex, costly workup needed to search for a primary tumor site of origin.[9]
Despite reports of precision medicine studies in pediatric oncology, none of them have described a case of cancer of unknown primary site with a defined or actionable genomic alteration.[10]
References
Kuttesch JF, Parham DM, Kaste SC, et al.: Embryonal malignancies of unknown primary origin in children. Cancer 75 (1): 115-21, 1995. [PUBMED Abstract]
Bohuslavizki KH, Klutmann S, Kröger S, et al.: FDG PET detection of unknown primary tumors. J Nucl Med 41 (5): 816-22, 2000. [PUBMED Abstract]
Han A, Xue J, Hu M, et al.: Clinical value of 18F-FDG PET-CT in detecting primary tumor for patients with carcinoma of unknown primary. Cancer Epidemiol 36 (5): 470-5, 2012. [PUBMED Abstract]
Tothill RW, Li J, Mileshkin L, et al.: Massively-parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. J Pathol 231 (4): 413-23, 2013. [PUBMED Abstract]
Varadhachary GR, Talantov D, Raber MN, et al.: Molecular profiling of carcinoma of unknown primary and correlation with clinical evaluation. J Clin Oncol 26 (27): 4442-8, 2008. [PUBMED Abstract]
Fizazi K, Greco FA, Pavlidis N, et al.: Cancers of unknown primary site: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 26 (Suppl 5): v133-8, 2015. [PUBMED Abstract]
Greco FA, Lennington WJ, Spigel DR, et al.: Poorly differentiated neoplasms of unknown primary site: diagnostic usefulness of a molecular cancer classifier assay. Mol Diagn Ther 19 (2): 91-7, 2015. [PUBMED Abstract]
Gatalica Z, Millis SZ, Vranic S, et al.: Comprehensive tumor profiling identifies numerous biomarkers of drug response in cancers of unknown primary site: analysis of 1806 cases. Oncotarget 5 (23): 12440-7, 2014. [PUBMED Abstract]
Ross JS, Wang K, Gay L, et al.: Comprehensive Genomic Profiling of Carcinoma of Unknown Primary Site: New Routes to Targeted Therapies. JAMA Oncol 1 (1): 40-9, 2015. [PUBMED Abstract]
Mody RJ, Prensner JR, Everett J, et al.: Precision medicine in pediatric oncology: Lessons learned and next steps. Pediatr Blood Cancer 64 (3): , 2017. [PUBMED Abstract]
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has slowly increased since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation to achieve optimal survival and quality of life:
Primary care physicians.
Pediatric surgeons.
Pathologists.
Pediatric radiation oncologists.
Pediatric medical oncologists and hematologists.
Ophthalmologists.
Rehabilitation specialists.
Pediatric oncology nurses.
Social workers.
Child-life professionals.
Psychologists.
Nutritionists.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children and adolescents with cancer.[2] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Other types of clinical trials test novel therapies when there is no standard therapy for a cancer diagnosis. Most of the progress in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[3–5] Childhood and adolescent cancer survivors require close monitoring because side effects of cancer therapy may persist or develop months or years after treatment. For information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
Childhood cancer is a rare disease, with about 15,000 cases diagnosed annually in the United States in individuals younger than 20 years.[6] The U.S. Rare Diseases Act of 2002 defines a rare disease as one that affects populations smaller than 200,000 people in the United States. Therefore, all pediatric cancers are considered rare.
The designation of a rare tumor is not uniform among pediatric and adult groups. In adults, rare cancers are defined as those with an annual incidence of fewer than six cases per 100,000 people. They account for up to 24% of all cancers diagnosed in the European Union and about 20% of all cancers diagnosed in the United States.[7,8] In children and adolescents, the designation of a rare tumor is not uniform among international groups, as follows:
A consensus effort between the European Union Joint Action on Rare Cancers and the European Cooperative Study Group for Rare Pediatric Cancers estimated that 11% of all cancers in patients younger than 20 years could be categorized as very rare. This consensus group defined very rare cancers as those with annual incidences of fewer than two cases per 1 million people. However, three additional histologies (thyroid carcinoma, melanoma, and testicular cancer) with incidences of more than two cases per 1 million people were also included in the very rare group due to a lack of knowledge and expertise in the management of these tumors.[9]
The Children’s Oncology Group defines rare pediatric cancers as those listed in the International Classification of Childhood Cancer subgroup XI, which includes thyroid cancers, melanomas and nonmelanoma skin cancers, and multiple types of carcinomas (e.g., adrenocortical carcinomas, nasopharyngeal carcinomas, and most adult-type carcinomas such as breast cancers and colorectal cancers).[10] These diagnoses account for about 5% of the cancers diagnosed in children aged 0 to 14 years and about 27% of the cancers diagnosed in adolescents aged 15 to 19 years.[4]
Most cancers in subgroup XI are either melanomas or thyroid cancers, with other cancer types accounting for only 2% of the cancers diagnosed in children aged 0 to 14 years and 9.3% of the cancers diagnosed in adolescents aged 15 to 19 years.
These rare cancers are extremely challenging to study because of the relatively few patients with any individual diagnosis, the predominance of rare cancers in the adolescent population, and the small number of clinical trials for adolescents with rare cancers.
Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010. [PUBMED Abstract]
American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed February 25, 2025.
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.
Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr. [PUBMED Abstract]
Gatta G, Capocaccia R, Botta L, et al.: Burden and centralised treatment in Europe of rare tumours: results of RARECAREnet-a population-based study. Lancet Oncol 18 (8): 1022-1039, 2017. [PUBMED Abstract]
DeSantis CE, Kramer JL, Jemal A: The burden of rare cancers in the United States. CA Cancer J Clin 67 (4): 261-272, 2017. [PUBMED Abstract]
Ferrari A, Brecht IB, Gatta G, et al.: Defining and listing very rare cancers of paediatric age: consensus of the Joint Action on Rare Cancers in cooperation with the European Cooperative Study Group for Pediatric Rare Tumors. Eur J Cancer 110: 120-126, 2019. [PUBMED Abstract]
Pappo AS, Krailo M, Chen Z, et al.: Infrequent tumor initiative of the Children’s Oncology Group: initial lessons learned and their impact on future plans. J Clin Oncol 28 (33): 5011-6, 2010. [PUBMED Abstract]
Treatment of Childhood Cancer of Unknown Primary
Chemotherapy, targeted therapy, and radiation therapy may be used to treat childhood cancers of unknown primary sites. The appropriate and relevant treatments, according to the general category of carcinoma or sarcoma (depending on the histological findings, symptoms, and extent of tumor), are initiated as early as possible.[1]
Morris GJ, Greco FA, Hainsworth JD, et al.: Cancer of unknown primary site. Semin Oncol 37 (2): 71-9, 2010. [PUBMED Abstract]
Treatment Options Under Clinical Evaluation for Childhood Cancer of Unknown Primary
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Latest Updates to This Summary (08/13/2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
This summary was comprehensively reviewed.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of pediatric cancer of unknown primary. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Cancer of Unknown Primary (CUP) Treatment are:
Denise Adams, MD (Children’s Hospital Boston)
Karen J. Marcus, MD, FACR (Dana-Farber of Boston Children’s Cancer Center and Blood Disorders Harvard Medical School)
William H. Meyer, MD
Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children’s Healthcare of Atlanta – Egleston Campus)
Alberto S. Pappo, MD (St. Jude Children’s Research Hospital)
Arthur Kim Ritchey, MD (Children’s Hospital of Pittsburgh of UPMC)
Carlos Rodriguez-Galindo, MD (St. Jude Children’s Research Hospital)
Stephen J. Shochat, MD (St. Jude Children’s Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Cancer of Unknown Primary (CUP) Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/unknown-primary/hp/child-unknown-primary-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 31909936]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Melanoma of the uveal tract (iris, ciliary body, and choroid) is rare, but it is the most common primary intraocular malignancy in adults. The mean age-adjusted incidence of uveal melanoma in the United States is approximately 4.3 new cases per million people, with no clear variation by latitude. The incidence is higher in men (4.9 cases per million) than in women (3.7 cases per million).[1] The age-adjusted incidence of this cancer has remained stable since at least the early 1970s.[1,2] U.S. incidence rates are lower than the rates of other reporting countries, which vary from about 5.3 to 10.9 cases per million. Some of the variation may be the result of differences in inclusion criteria and methods of calculation.[1]
Uveal melanoma is most often diagnosed in older individuals, with a progressively rising, age-specific incidence rate that peaks near age 70 years.[3]
Host susceptibility factors associated with the development of this cancer include:[2–4]
White race and ethnicity.
Light eye color.
Fair skin.
The ability to tan.
In view of these susceptibility factors, numerous observational studies have explored the relationship between sunlight exposure and risk of uveal melanoma. These studies have found only weak associations or yielded contradictory results.[3] Similarly, there is no consistent evidence that occupational exposure to UV light or other agents is a risk factor for uveal melanoma.[3,5]
Anatomy
Uveal melanomas can arise in the anterior (iris) or the posterior (ciliary body or choroid) uveal tract.[6] Most uveal tract melanomas originate in the choroid. The ciliary body is a less common site of origin, and the iris is the least common. The comparatively low incidence of iris melanomas has been attributed to the characteristic features of these tumors; they tend to be smaller, slower growing, and relatively dormant compared with their posterior counterparts. Iris melanomas rarely metastasize.[7] Melanomas of the posterior uveal tract generally have a more malignant histological appearance; are detected later; and metastasize more frequently than iris melanomas. The typical choroidal melanoma is a brown, elevated, dome-shaped subretinal mass. The degree of pigmentation ranges from dark brown to totally amelanotic.
Most uveal melanomas are initially completely asymptomatic. As the tumor enlarges, it may cause distortion of the pupil (iris melanoma), blurred vision (ciliary body melanoma), or markedly decreased visual acuity caused by secondary retinal detachment (choroidal melanoma). Serous detachment of the retina may occur. If extensive detachment occurs, secondary angle-closure glaucoma occasionally develops. Clinically, several lesions simulate uveal melanoma, including metastatic carcinoma, posterior scleritis, and benign tumors, such as nevi and hemangiomas.[8]
Careful examination by an experienced clinician remains the most important test to diagnose intraocular melanoma. A small uveal melanoma cannot be distinguished from a nevus. Small uveal lesions are observed for growth before making a diagnosis of melanoma. Clinical findings that may help to identify melanoma include:[6]
Orange pigment on the tumor surface.
Subretinal fluid.
Tumor thickness of more than 2 mm.
Low internal reflectivity on ultrasound examination.
Ancillary diagnostic testing, including fluorescein angiography and ultrasonography, can be extremely valuable in establishing and confirming the diagnosis.[9] In a large, retrospective, single-center series of 2,514 consecutive patients with choroidal nevi, the progression rate to melanoma was 8.6% at 5 years, 12.8% at 10 years, and 17.3% at 15 years.[10]
Prognostic Factors
Several factors influence prognosis. The most important factors include:
Several additional microscopic features can affect the prognosis of intraocular melanoma, including:
Mitotic activity.
Lymphocytic infiltration.
Fibrovascular loops (possibly).
Cell type is the most commonly used predictor of outcome following enucleation. Patients with spindle-A cell melanomas have the best prognosis and patients with epithelioid cell melanomas have the least favorable prognosis.[1,4,9] Nevertheless, most tumors have an admixture of cell types, and there is no clear consensus regarding the proportion of epithelioid cells that constitutes designation of a tumor as mixed or epithelioid.[6]
Extraocular extension, recurrence, and metastasis are associated with an extremely poor prognosis, and long-term survival cannot be expected for patients with these features.[11] The 5-year mortality rate for patients with metastasis from ciliary body or choroidal melanoma is approximately 30%, compared with a rate of 2% to 3% for patients with iris melanomas.[12]
References
Singh AD, Topham A: Incidence of uveal melanoma in the United States: 1973-1997. Ophthalmology 110 (5): 956-61, 2003. [PUBMED Abstract]
Inskip PD, Devesa SS, Fraumeni JF: Trends in the incidence of ocular melanoma in the United States, 1974-1998. Cancer Causes Control 14 (3): 251-7, 2003. [PUBMED Abstract]
Weis E, Shah CP, Lajous M, et al.: The association between host susceptibility factors and uveal melanoma: a meta-analysis. Arch Ophthalmol 124 (1): 54-60, 2006. [PUBMED Abstract]
Harris RB, Griffith K, Moon TE: Trends in the incidence of nonmelanoma skin cancers in southeastern Arizona, 1985-1996. J Am Acad Dermatol 45 (4): 528-36, 2001. [PUBMED Abstract]
Albert DM, Kulkarni AD: Intraocular melanoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 2090-8.
Shields CL, Furuta M, Berman EL, et al.: Choroidal nevus transformation into melanoma: analysis of 2514 consecutive cases. Arch Ophthalmol 127 (8): 981-7, 2009. [PUBMED Abstract]
Gragoudas ES, Egan KM, Seddon JM, et al.: Survival of patients with metastases from uveal melanoma. Ophthalmology 98 (3): 383-9; discussion 390, 1991. [PUBMED Abstract]
Introduction to melanocytic tumors of the uvea. In: Shields JA, Shields CL: Intraocular Tumors: A Text and Atlas. Saunders, 1992, pp 45-59.
Cellular Classification of Intraocular (Uveal) Melanoma
Primary intraocular melanomas originate from melanocytes in the uveal tract.[1] The following four distinct cellular types are recognized in intraocular melanoma (revised Callender classification):[2]
Spindle-A cells (spindle-shaped cells with slender nuclei and lacking visible nucleoli).
Spindle-B cells (spindle-shaped cells with larger nuclei and distinct nucleoli).
Epithelioid cells (larger polygonal cells with one or more prominent nucleoli).
Intermediate cells (similar to but smaller than epithelioid cells).
Most primary intraocular melanomas contain variable proportions of epithelioid, spindle-A, and spindle-B cells (mixed-cell melanomas). Pure epithelioid-cell primary melanomas are infrequent (approximately 3% of cases).[1] In the Collaborative Ocular Melanoma Study, mixed-cell melanomas predominated (86% of cases).[3]
References
Klintworth GK, Scroggs MW: The eye and ocular adnexa. In: Sternberg SS, ed.: Diagnostic Surgical Pathology. Lippincott Williams & Wilkins, 1999, pp 994-6.
Grossniklaus HE, Green WR: Uveal tumors. In: Garner A, Klintworth GK, eds.: Pathobiology of Ocular Disease: A Dynamic Approach. 2nd ed. M. Dekker, 1994, pp 1423-77.
Histopathologic characteristics of uveal melanomas in eyes enucleated from the Collaborative Ocular Melanoma Study. COMS report no. 6. Am J Ophthalmol 125 (6): 745-66, 1998. [PUBMED Abstract]
Classification and Stage Information for Intraocular (Uveal) Melanoma
Tumor Size
Uveal melanoma most often assumes a nodular or dome-shaped configuration. Occasionally, tumors are flat or diffuse and involve extensive areas of the uvea with little elevation.
Tumor size classifications according to boundary lines used in a Collaborative Ocular Melanoma Study (COMS) are as follows:[1]
Small: Range from 1.0 to 3.0 mm in apical height and largest basal diameter of 5.0 to 16.0 mm.[1]
Medium: Range from 3.1 to 8.0 mm in apical height and a basal diameter of not more than 16.0 mm.[2]
Large: Greater than 8.0 mm in apical height or a basal diameter more than 16.0 mm, when the apical height is at least 2.0 mm.
Although most ocular melanomas have a raised configuration, about 5% grow in a diffuse pattern that also may have prognostic significance. The tumors have a horizontal, flat-growth pattern, with the thickness measuring approximately 20% or less than the greatest basal dimension. This uncommon variant of uveal melanoma seems to be associated with a poorer prognosis, particularly when the diameter is large and the margins are poorly defined.[3]
In clinical practice, the tumor base may be estimated in average optic disc diameters (1 dd = 1.5 mm). The average elevation may be estimated in diopters (3 diopters = 1 mm). Other techniques, such as ultrasonography, are used to provide more accurate measurements.
An important function of ophthalmic ultrasonography is the detection of extrascleral extension.[4,5] Extrascleral extension measuring 2 mm or more in thickness can be demonstrated, provided it is located behind the equator where the intraocular tumor, sclera, and adjacent orbital fat are readily imaged.[6] Orbital extraocular extension of choroidal melanoma may be found in eyes with medium and large tumors, but it is very rare in eyes with small melanomas.
Metastatic Disease
Systemic metastases are evident in only 2% to 3% of patients at the time of diagnosis of the primary ocular melanoma.[7] Because the uveal tract is a vascular structure without lymphatic channels, tumor spread occurs principally by local extension and by dissemination through the bloodstream.[7] Lymphatic spread is rare but may occur after local extension into the conjunctiva and its lymphatics.[8] Given the rarity of nodal metastases, sentinel node biopsies of nonclinically involved nodes are not done as part of the staging procedure.[7]
Systemic metastases are generally hematogenous in origin, and the first site identified is usually the liver.[9] Lung, bone, and subcutaneous sites are also common.[9] In the COMS trials, the liver was the only site of detectable metastasis in 46% of patients with metastases reported during follow-up or at the time of death; 43% had metastases diagnosed in the liver and other sites.[9] In patients with a history of ocular melanoma who present with hepatic metastases of unknown origin, metastatic melanoma is considered in the differential diagnosis.
It is particularly unusual for choroidal melanomas of any size to invade the optic nerve or its meninges.[10] Metastasis of choroidal melanoma to the contralateral choroid is also rare.[9,11]
Staging
American Joint Committee on Cancer (AJCC) stage groupings and definitions of TNM
The AJCC has designated staging by TNM (tumor, node, metastasis) classification to define melanoma of the uveal tract.[7]
As in the seventh edition of the AJCC Cancer Staging Manual, there is no staging system for iris melanomas in the eighth edition. However, TNM should still be recorded for this site and histology combination.
Table 1. Definition of Primary Tumor (T) for Iris Melanomasa,b
T Category
T Criteria
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
bIris melanomas originate from, and are predominantly located in, this region of the uvea. If less than half the tumor volume is located within the iris, the tumor may have originated in the ciliary body, and consideration should be given to classifying it accordingly.
TX
Primary tumor cannot be assessed.
T0
No evidence of primary tumor.
T1
Tumor limited to the iris.
–T1a
Tumor limited to the iris, not more than 3 clock hours in size.
–T1b
Tumor limited to the iris, more than 3 clock hours in size.
–T1c
Tumor limited to the iris with secondary glaucoma.
T2
Tumor confluent with or extending into the ciliary body, choroid, or both.
–T2a
Tumor confluent with or extending into the ciliary body, without secondary glaucoma.
–T2b
Tumor confluent with or extending into the ciliary body and choroid, without secondary glaucoma.
–T2c
Tumor confluent with or extending into the ciliary body, choroid, or both, with secondary glaucoma.
T3
Tumor confluent with or extending into the ciliary body, choroid, or both, with scleral extension.
T4
Tumor with extrascleral extension.
–T4a
Tumor with extrascleral extension ≤5 mm in largest diameter.
–T4b
Tumor with extrascleral extension >5 mm in largest diameter.
Table 2. Definition of Regional Lymph Node (N)a
N Category
N Criteria
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
NX
Regional lymph nodes cannot be assessed.
N0
No regional lymph node involvement.
N1
Regional lymph node metastases or discrete tumor deposits in the orbit.
–N1a
Metastasis in one or more regional lymph node(s).
–N1b
No regional lymph nodes are positive, but there are discrete tumor deposits in the orbit that are not contiguous to the eye (choroidal and ciliary body).
Table 3. Definition of Distant Metastasis (M)a
M Category
M Criteria
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
M0
No distant metastasis by clinical classification.
M1
Distant metastasis.
–M1a
Largest diameter of the largest metastasis ≤3.0 cm.
–M1b
Largest diameter of the largest metastasis 3.1–8.0 cm.
–M1c
Largest diameter of the largest metastasis ≥8.1 cm.
Table 4. Classification of Ciliary Body and Choroid Uveal Melanoma Based on Thickness and Diametera
Category
Tumor Size
aAdapted from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
1
Tumor is ≤12 mm in diameter and ≤3 mm in thickness; or
Tumor is ≤9 mm in diameter and 3.1–6 mm in thickness.
2
Tumor is 12.1–18 mm in diameter and ≤3 mm in thickness; or
Tumor is 9.1–15 mm in diameter and 3.1– 6 mm in thickness; or
Tumor is ≤12 mm in diameter and 6.1–9 mm in thickness.
3
Tumor is 15.1–18 mm in diameter and 3.1–6 mm in thickness; or
Tumor is 12.1–18 mm in diameter and 6.1–9 mm in thickness; or
Tumor is ≤18 mm in diameter and 9.1–12 mm in thickness; or
Tumor is ≤15 mm in diameter and 12.1–15 mm in thickness.
4
Tumor is >18 mm in diameter and may be any thickness; or
Tumor is 15.1–18 mm in diameter and >12 mm in thickness; or
Tumor is ≤15 mm in diameter and >15 mm in thickness.
Table 5. Definition of TNM Stage I Choroidal and Ciliary Body Melanomasa,b
Stage
TNM
Description
M = distant metastasis; N = regional lymph node; T = primary tumor.
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
b1) Primary ciliary body and choroidal melanomas are classified according to four tumor-size categories based on thickness and diameter. See Table 4. 2) In clinical practice, the largest tumor basal diameter may be estimated in optic disc diameters (DD) (average: 1 DD = 1.5 mm), and tumor thickness may be estimated in diopters (average: 2.5 diopters = 1 mm). Ultrasonography and fundus photography are used to provide more accurate measurements. 3) When histopathological measurements are recorded after fixation, tumor diameter and thickness may be underestimated because of tissue shrinkage.
I
T1a, N0, M0
–T1a = Tumor size category 1 without ciliary body involvement and extraocular extension.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
Table 6. Definition of TNM Stages IIA and IIB Choroidal and Ciliary Body Melanomasa,b
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis.
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
b1) Primary ciliary body and choroidal melanomas are classified according to four tumor-size categories based on thickness and diameter. See Table 4. 2) In clinical practice, the largest tumor basal diameter may be estimated in optic disc diameters (DD) (average: 1 DD = 1.5 mm), and tumor thickness may be estimated in diopters (average: 2.5 diopters = 1 mm). Ultrasonography and fundus photography are used to provide more accurate measurements. 3) When histopathological measurements are recorded after fixation, tumor diameter and thickness may be underestimated because of tissue shrinkage.
IIA
T1b–d, N0, M0
–T1b = Tumor size category 1 with ciliary body involvement.
–T1c = Tumor size category 1 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T1d = Tumor size category 1 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T2a, N0, M0
–T2a = Tumor size category 2 without ciliary body involvement and extraocular extension.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
IIB
T2b, N0, M0
–T2b = Tumor size category 2 with ciliary body involvement.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T3a, N0, M0
–T3a = Tumor size category 3 without ciliary body involvement and extraocular extension.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
Table 7. Definition of TNM Stages IIIA, IIIB, and IIIC Choroidal and Ciliary Body Melanomasa,b
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis.
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
b1) Primary ciliary body and choroidal melanomas are classified according to four tumor-size categories based on thickness and diameter. See Table 4. 2) In clinical practice, the largest tumor basal diameter may be estimated in optic disc diameters (DD) (average: 1 DD = 1.5 mm), and tumor thickness may be estimated in diopters (average: 2.5 diopters = 1 mm). Ultrasonography and fundus photography are used to provide more accurate measurements. 3) When histopathological measurements are recorded after fixation, tumor diameter and thickness may be underestimated because of tissue shrinkage.
IIIA
T2c–d, N0, M0
–T2c = Tumor size category 2 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T2d = Tumor size category 2 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T3b–c, N0, M0
–T3b = Tumor size category 3 with ciliary body involvement.
–T3c = Tumor size category 3 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T4a, N0, M0
–T4a = Tumor size category 4 without ciliary body involvement and extraocular extension.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
IIIB
T3d, N0, M0
–T3d = Tumor size category 3 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T4b–c, N0, M0
–T4b = Tumor size category 4 with ciliary body involvement.
–T4c = Tumor size category 4 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
IIIC
T4d–e, N0, M0
–T4d = Tumor size category 4 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
–T4e = Any tumor size category with extraocular extension >5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
Table 8. Definition of TNM Stage IV Choroidal and Ciliary Body Melanomasa,b
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis.
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
b1) Primary ciliary body and choroidal melanomas are classified according to four tumor-size categories based on thickness and diameter. See Table 4. 2) In clinical practice, the largest tumor basal diameter may be estimated in optic disc diameters (DD) (average: 1 DD = 1.5 mm), and tumor thickness may be estimated in diopters (average: 2.5 diopters = 1 mm). Ultrasonography and fundus photography are used to provide more accurate measurements. 3) When histopathological measurements are recorded after fixation, tumor diameter and thickness may be underestimated because of tissue shrinkage.
IV
Any T, N1, M0
TX = Primary tumor cannot be assessed.
T0 = No evidence of primary tumor.
T1 = Tumor size category 1.
–T1a = Tumor size category 1 without ciliary body involvement and extraocular extension.
–T1b = Tumor size category 1 with ciliary body involvement.
–T1c = Tumor size category 1 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T1d = Tumor size category 1 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
T2 = Tumor size category 2.
–T2a = Tumor size category 2 without ciliary body involvement and extraocular extension.
–T2b = Tumor size category 2 with ciliary body involvement.
–T2c = Tumor size category 2 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T2d = Tumor size category 2 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
T3 = Tumor size category 3.
–T3a = Tumor size category 3 without ciliary body involvement and extraocular extension.
–T3b = Tumor size category 3 with ciliary body involvement.
–T3c = Tumor size category 3 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T3d = Tumor size category 3 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
T4 = Tumor size category 4.
–T4a = Tumor size category 4 without ciliary body involvement and extraocular extension.
–T4b = Tumor size category 4 with ciliary body involvement.
–T4c = Tumor size category 4 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T4d = Tumor size category 4 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
–T4e = Any tumor size category with extraocular extension >5 mm in largest diameter.
N1 = Regional lymph node metastases or discrete tumor deposits in the orbit.
–N1a = Metastasis in one or more regional lymph nodes(s).
–N1b = No regional lymph nodes are positive, but there are discrete tumor deposits in the orbit that are not contiguous to the eye.
M0 = No distant metastasis by clinical classification.
Any T, Any N, M1a–c
Any T = See descriptions above in this table, stage IV, Any T, N1, M0.
NX = Regional lymph nodes cannot be assessed.
N0 = No regional lymph node involvement.
N1 = Regional lymph node metastases or discrete tumor deposits in the orbit.
–N1a = Metastasis in one or more regional lymph node(s).
–N1b = No regional lymph nodes are positive, but there are discrete tumor deposits in the orbit that are not contiguous to the eye (choroidal and ciliary body).
M1 = Distant metastasis.
–M1a = Largest diameter of the largest metastasis ≤3.0 cm.
–M1b = Largest diameter of the largest metastasis 3.1–8.0 cm.
–M1c = Largest diameter of the largest metastasis ≥8.1 cm.
Prognostic features
There are several key prognostic features that are important to collect in malignant melanoma of the uvea, even though they are not included in staging algorithms. These include:[7]
Molecular features
Chromosomal alterations.
Chromosome 3 status (loss or no loss, complete or partial).
Chromosome 6p status (gain or no gain).
Chromosome 8q status (gain or no gain).
Indicate:
Technique used for assessing chromosome status may include:
Karyotyping.
Fluorescence in situ hybridization.
Comparative genomic hybridization.
Loss of heterozygosity using DNA polymorphism analysis (e.g., single nucleotide polymorphism, microsatellite).
Other.
How specimen was obtained may include:
Enucleation.
Local resection.
Biopsy.
Fine-needle aspiration biopsy.
For needle biopsies, whether cytopathological evaluation was performed to confirm the presence of tumor cells.
Gene-expression profile: class 1 or class 2.
Indicate:
Technique used for gene-expression profiling may include:
Microarray.
Pathological complete response.
How specimen was obtained may include:
Enucleation.
Local resection.
Biopsy.
Fine-needle aspiration biopsy.
For needle biopsies, whether cytopathological evaluation was performed to confirm the presence of tumor cells.
Clinical and histopathological features
Clinical.
Positron emission tomography/computed tomography.
Fluorine F 18-fludeoxyglucose standardized uptake values (higher values in primary tumor may be associated with shorter survival).
Confocal indocyanine green angiography.
Identification of complex monocirculatory patterns (i.e., loops, networks, arcs with branching, parallel with cross-linking or a combination of these patterns may be associated with shorter survival).
Histopathological.
Mitotic count.
Number of mitotic figures per 40 high-power fields (typical field area 0.15–0.19 mm2, higher counts are associated with shorter survival).
Mean diameter of the ten largest nucleoli.
Mean of the longest nucleoli (MLN) is measured along a central 5-mm long strip, e.g., after silver staining (larger values are associated with shorter survival).
Presence of extravascular matrix patterns.
Loops.
Absent.
Present (shorter survival).
Loops forming networks.
Absent.
Present (shorter survival).
Other complex patterns (arcs with branching, parallel with cross-linking; absent or present).
The patterns are assessed with light microscopy under a dark green filter after staining with periodic-acid Schiff without counterstain.
Microvascular density.
Number of immunopositive elements labeled with markers for vascular endothelial cells (e.g., CD34 epitope, factor VIII-related antigen) in areas of densest vascularization (typical field area 0.31 mm2, higher counts are associated with shorter survival).
Insulin-like growth factor 1 receptor (IGF1-R).
Percentage of immunopositive tumor cells (high expression is associated with shorter survival).
Tumor-infiltrating lymphocytes.
Few (longest survival).
Moderate numbers.
Many (shortest survival).
Tumor-infiltrating macrophages.
Few (longest survival).
Moderate numbers.
Many (shortest survival).
The number can be compared with standard photographs.[12]
HLA class I expression.
Percentage of immunopositive tumor cells (low expression is associated with longer survival).
References
Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 115 (12): 1537-44, 1997. [PUBMED Abstract]
Diener-West M, Earle JD, Fine SL, et al.: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, II: characteristics of patients enrolled and not enrolled. COMS Report No. 17. Arch Ophthalmol 119 (7): 951-65, 2001. [PUBMED Abstract]
Shields CL, Shields JA, De Potter P, et al.: Diffuse choroidal melanoma. Clinical features predictive of metastasis. Arch Ophthalmol 114 (8): 956-63, 1996. [PUBMED Abstract]
Scott IU, Murray TG, Hughes JR: Evaluation of imaging techniques for detection of extraocular extension of choroidal melanoma. Arch Ophthalmol 116 (7): 897-9, 1998. [PUBMED Abstract]
Romero JM, Finger PT, Iezzi R, et al.: Three-dimensional ultrasonography of choroidal melanoma: extrascleral extension. Am J Ophthalmol 126 (6): 842-4, 1998. [PUBMED Abstract]
Echography (ultrasound) procedures for the Collaborative Ocular Melanoma Study (COMS), Report no. 12, Part I. J Ophthalmic Nurs Technol 18 (4): 143-9, 1999 Jul-Aug. [PUBMED Abstract]
Dithmar S, Diaz CE, Grossniklaus HE: Intraocular melanoma spread to regional lymph nodes: report of two cases. Retina 20 (1): 76-9, 2000. [PUBMED Abstract]
Diener-West M, Reynolds SM, Agugliaro DJ, et al.: Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol 123 (12): 1639-43, 2005. [PUBMED Abstract]
Shields CL, Santos MC, Shields JA, et al.: Extraocular extension of unrecognized choroidal melanoma simulating a primary optic nerve tumor: report of two cases. Ophthalmology 106 (7): 1349-52, 1999. [PUBMED Abstract]
Singh AD, Shields JA, Shields CL, et al.: Choroidal melanoma metastatic to the contralateral choroid. Am J Ophthalmol 132 (6): 941-3, 2001. [PUBMED Abstract]
Mäkitie T, Summanen P, Tarkkanen A, et al.: Tumor-infiltrating macrophages (CD68(+) cells) and prognosis in malignant uveal melanoma. Invest Ophthalmol Vis Sci 42 (7): 1414-21, 2001. [PUBMED Abstract]
Treatment Option Overview for Intraocular (Uveal) Melanoma
Observation
Patients with iris melanomas have relatively good outcomes, with 5-year survival rates exceeding 95%. Iris melanomas are predominantly of the spindle-cell type and are usually smaller in size than posterior melanomas because they are detected earlier. Conservative management is generally advocated whenever possible, but surgical intervention may be justified with unequivocal tumor growth or with extensive disease at initial examination.
The management of small choroidal melanomas is controversial, and it is not clear whether treating small tumors prevents metastasis.[1] The natural history of small choroidal melanomas is poorly understood. Small, pigmented, choroidal lesions cannot always be differentiated reliably on examination. Growth is a presumed indicator of malignant potential.[2] The likelihood of progression from the time of diagnosis to the time when tumor growth warrants treatment has not been well characterized. Some ophthalmologists advocate observation. This course has been justified on several grounds, including the difficulty of establishing a correct diagnosis, the lack of any documented efficacy for globe-conserving treatments, and concerns for severe treatment-related morbidity. Others have advocated earlier therapeutic intervention.[1,3,4]
Although patients diagnosed with small choroidal tumors were not eligible for participation in the Collaborative Ocular Melanoma Study (COMS), these patients could participate in a prospective follow-up study to evaluate the natural history of small lesions. The 2-year tumor growth estimates were 21%, and the 5-year tumor growth estimates were 31%.[5] Clinical risk factors associated with tumor growth included:[3,5]
Increased tumor thickness.
Presence of subretinal fluid.
Orange pigmentation.
Absence of drusen.
Absence of retinal pigment.
Margin at the optic disc.
Epithelial changes surrounding the tumor.
Surgery
The selection of treatment depends on the following:
Site of origin (choroid, ciliary body, or iris).
Size and location of the lesion.
Age of the patient.
Occurrence of extraocular invasion, recurrence, or metastasis.
Enucleation
In the past, enucleation (eye removal) was the standard treatment for primary choroidal melanoma, and it is still used when large tumors are present. However, enucleation has been largely replaced by radiation therapy (i.e., brachytherapy with radioactive plaques or external-beam, charged-particle radiation therapy) to spare the affected eye.[6,7]
A randomized trial evaluated the role of pre-enucleation EBRT for large choroidal tumors that required enucleation. The study found that EBRT had no impact on overall survival (OS).[8,9][Level of evidence A1] A COMS trial included 1,003 patients with large choroidal melanomas (≥2 mm in height and ≥16 mm in diameter, or ≥10 mm in height irrespective of diameter, or ≥8 mm in height and border <2 mm from the optic disc) with no known metastases. Patients were randomly assigned to receive enucleation alone or enucleation after preoperative external photon-beam radiation from cobalt 60 or accelerators (20 Gy in 5 daily fractions) to the orbit and globe.[8,9] Through 10 years of follow-up, the median survival in both arms was approximately 7 years, and the 10-year all-cause mortality rate was 61% in both arms (relative risk [RR]death, 1.00; 95% confidence interval [CI], 0.85–1.18). Metastasis-free survival was also nearly identical in both arms.
Transscleral local resection
Eye-sparing transscleral local resection plays a limited role in the management of uveal melanoma. It is used in patients with large choroidal and ciliary body tumors who are not candidates for radiation therapy but are highly motivated to retain their eye.[10–12] The procedure is technically demanding and is generally performed only in centers with specialized expertise in this surgery. There is a substantial risk of retinal detachment, intraocular bleeding, and complications associated with the anesthesia-induced hypotension used to decrease the risk of bleeding. Either adjuvant brachytherapy or neoadjuvant proton-beam therapy are administered. Experience is limited to retrospective, single-center, case series.[10–12][Level of evidence C3]
Surgical resection of metastases
Surgical resection of metastases from ocular melanoma has been reported in case series of highly selected patients with occasional favorable outcomes.[13,14] However, the favorable outcomes may be the result of strong patient-selection factors, and the role of resection in this setting is unclear.[13,14][Level of evidence C3]
Radiation Therapy
Episcleral brachytherapy using plaques containing small radioactive seeds is the most common form of radiation used in the management of intraocular melanoma. Iodine I 125 (125I), cobalt Co 60 (60Co), palladium Pd 103 (103Pd), iridium Ir 192 (192Ir), and ruthenium Ru 106 (106Ru) are examples of radioactive isotopes used in the brachytherapy plaques. Isotopes with relatively low photon and electron emissions (125I, 103Pd, and 106Ru) are more easily shielded to reduce the exposure to adjacent normal tissues, and 125I is probably the most commonly used radioisotope.[15] Although plaque radiation therapy allows preservation of the eye, visual acuity is frequently lost over time.
In a case series of 1,106 patients who were treated with plaque radiation therapy for uveal melanoma and who had an initial acuity of at least 20/100, 68% developed poor acuity (i.e., 20/200 or worse) within 10 years.[16]
Factors associated with worse acuity outcomes included:[16]
Age older than 60 years.
Diminished baseline acuity.
Diabetes.
Increased tumor size and thickness.
Location near the fovea or optic disc.
Type of isotope therapy (106Ru, 60Co, or 192Ir vs. 125I).
125I brachytherapy yields equivalent overall and melanoma metastasis-specific survival rates to enucleation for medium-sized melanomas.[17][Level of evidence A1] The randomized COMS Medium Tumor Trial compared 125I episcleral-plaque brachytherapy (85 Gy at 0.42–1.06 Gy/hr) with enucleation in 1,317 patients with medium-sized choroidal tumors (tumor height 2.5–10.0 mm and tumor diameter ≤16.0 mm that were not contiguous with the optic disc).[17] Eighty-five percent of the patients treated with 125I brachytherapy retained their eye for 5 years or more, and 37% of them had visual acuity better than 20/200 in the irradiated eye 5 years after treatment.[17] No statistically significant differences in mortality were observed between the two study arms after 12 years of follow-up, either for death from all causes or death with histopathologically confirmed melanoma metastasis.[18] The 5- and 10-year all-cause mortality rates were 19% and 35% in both study arms. The cumulative all-cause mortality rate at 12 years was 43% in the 125I arm versus 41% in the enucleation arm (RR, 1.04; 95% CI, 0.86–1.24). The 5-year metastasis-specific mortality rates were 13% in both arms; at 10 years, the rates were 21% and 22% (RR for metastasis-specific mortality, 1.07; 95% CI, 0.81–1.41 through 12 years).
In a companion study within the COMS, 209 patients were prospectively assessed for quality of life during the first 5 years of follow-up.[19] Both study groups reported increasing difficulty with vision-oriented daily activities and ocular pain as time elapsed. Most measures of visual function were similar between the two groups. However, there were statistically significant differences favoring the brachytherapy group in comfort with driving for the first year after therapy and in reported peripheral vision for the first 2 years after therapy. These differences disappeared by year 5 of follow-up.[19][Level of evidence A3]
Charged-particle EBRT (using protons, carbon ions, or helium ions) is the other major form of radiation therapy used in the management of ocular melanomas.[20–23] This form of radiation therapy requires sophisticated equipment available only at selected centers. In addition, charged-particle EBRT involves patient cooperation during treatment (e.g., voluntarily fixating the eye on a particular point so the tumor is positioned properly in the radiation beam). A lower risk of early and late local radiation failures has been reported after charged-particle EBRT than after the use of brachytherapy, possibly resulting from differences in dose distribution in the two techniques.[20][Levels of evidence B3 and C3]
In a single-center, single-surgeon study, 184 patients with uveal melanomas smaller than 15 mm in diameter and smaller than 10 mm in thickness were randomly assigned to receive 125I brachytherapy versus helium ion radiation (to an estimated dose of 70 Gy equivalents in 5 fractions over 7 to 11 days in each arm).[24] The local tumor regrowth rate by 4 years was 13.3% in the brachytherapy arm compared with 0% in the helium ion arm (P < .001). However, the rates of metastasis, death from metastasis, and overall mortality were similar in both arms.[24][Level of evidence B3]
Because of its dose distribution, charged-particle irradiation can be better used than plaque brachytherapy to treat larger tumors and tumors closer to the fovea or optic disc. A large, single-center, single-surgeon series of 2,069 patients treated with proton-beam therapy had an actuarial local control rate of 95% (95% CI, 93%–96%) at 15 years. The cumulative rate of enucleation was 16% (95% CI, 13%–20%). Enucleations were primarily the result of neovascular glaucoma (46% of enucleations), blind uncomfortable eyes (31% of enucleations), or local recurrence (23% of enucleations). As with plaque radiation, risk factors for deterioration in visual acuity after charged-particle radiation were tumor size, location near the fovea or optic disc, baseline acuity, and underlying diabetes.[21]
Similarly, another large, single-center, single-surgeon, consecutive series of 886 patients treated with proton-beam irradiation reported a local control rate of 92.1% (95% CI, 89.8%–94.6%) and ocular conservation rate of 87.3% (95% CI, 83.9%–90.9%) at 10 years.[22][Level of evidence C3] The actuarial OS rate at 10 years was 64.1% (95% CI, 59.5%–69.0%).
In a single-center, phase I/II study of 57 evaluable patients treated with carbon ion-beam irradiation and followed for a median of 26 months, 26 patients developed neovascular glaucoma or severe eye pain from increased intraocular pressure, and 3 patients underwent enucleation. One patient had a local tumor recurrence.[23]
In an attempt to lower the complication rate and improve functional outcomes, one study compared a decreased dose of 50 cobalt Gy equivalents (CGE) with 70 CGE proton beam (each delivered in 5 fractions, usually within a 7-day period). Patients (n = 188) with tumors smaller than 15 mm in diameter and smaller than 5 mm in height, which were located near the optic disc or macula, were randomly assigned to the two doses in a double-masked study design. At 5 years, there were no statistically significant differences in local tumor control, rate of metastasis, visual acuity, or complication rates. However, the visual fields were better in the 50-CGE group.[25][Level of evidence B3]
As noted in the Surgery section, the role of pre-enucleation external photon-beam radiation therapy has been tested in a randomized trial and has shown no impact on OS for large choroidal tumors treated with enucleation.[8,9]
External-beam–photon-beam (gamma-ray) radiation therapy with gamma-knife stereotactic radiation surgery as a single-fraction [26] or fractionated stereotactic radiation [27,28] is being investigated as an alternative to brachytherapy or charged-beam radiation for posterior uveal melanomas, particularly for tumors too large or too close to the optic disc or macula to treat with brachytherapy. Because the dose rate of radiation has a slower delivery time than is the case with charged particles, specialized techniques are used to immobilize the eye [26] or to avoid delivery of the photons while the eye is moving or closed.[28] Experience is more limited with external-beam–photon therapy than for either brachytherapy or charged-particle EBRT, and there are no controlled comparisons with either of the other techniques. Early results from single-center series suggest similar levels of local tumor control and eye retention rates, but patient-selection factors may play a role.[28][Level of evidence C3]
Transpupillary Thermotherapy
Transpupillary thermotherapy (TTT) directs an infrared laser, usually at a wavelength of 810 nm, through a dilated pupil in one or more sessions to induce heat necrosis of uveal melanomas. This method carries the theoretical advantage of high-precision destruction of tumor tissue under direct visualization. However, TTT has important limitations that confine its use to specific circumstances.[1,29] The limited ability of TTT to penetrate thick tumors with sufficient energy restricts its use to small melanomas or tumors of a size that some ophthalmologists recommend for follow-up without any initial therapy. For more information, see the Observation section. When used as the primary therapy, there are relatively high rates of local recurrence and retinal vascular damage. Recurrence rates are particularly high when the tumor abuts the optic nerve and overhangs the optic disc.[1][Level of evidence C3]
In a single-center study, 95 patients with small choroidal melanomas (diameter <10 mm and thickness <3.5 mm) were randomly assigned to receive either TTT or 125I brachytherapy (100 Gy).[30] The tumor regression rate was 92% in the TTT arm and 98% in the 125I arm (P = .4). With a mean follow-up time of 56.2 months, there were four recurrences in the TTT arm and one in the 125I arm. However, the study was too small to provide clear information on efficacy differences.
TTT is also under evaluation as an adjunct to primary therapy with proton-beam radiation. In large uveal melanomas, proton-beam therapy is associated with exudative, inflammatory, and glaucomatous complications that may require enucleation. In a single-center trial, 151 patients with uveal melanomas at least 7 mm thick or at least 15 mm in diameter were randomly assigned to receive proton-beam radiation (60 CGEs over four daily fractions) with or without TTT (810 nm wavelength at 1, 6, and 12 months after therapy) and followed for a median of 38 months.[31] There were no differences between the two groups in maculopathy, papillopathy, or glaucoma. The enucleation rate was lower in the TTT group (about 2% vs. 18% at 5 years, P = .02). However, the study was not masked, and replication of the results would be important.
There are uncertainties at all stages about the optimal management of intraocular melanoma. Physicians should discuss clinical trial opportunities with eligible patients. Information about ongoing clinical trials is available from the NCI website.
References
Shields CL, Shields JA, Perez N, et al.: Primary transpupillary thermotherapy for small choroidal melanoma in 256 consecutive cases: outcomes and limitations. Ophthalmology 109 (2): 225-34, 2002. [PUBMED Abstract]
Augsburger JJ: Is observation really appropriate for small choroidal melanomas. Trans Am Ophthalmol Soc 91: 147-68; discussion 169-75, 1993. [PUBMED Abstract]
Shields CL, Cater J, Shields JA, et al.: Combination of clinical factors predictive of growth of small choroidal melanocytic tumors. Arch Ophthalmol 118 (3): 360-4, 2000. [PUBMED Abstract]
Robertson DM, Buettner H, Bennett SR: Transpupillary thermotherapy as primary treatment for small choroidal melanomas. Arch Ophthalmol 117 (11): 1512-9, 1999. [PUBMED Abstract]
Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 115 (12): 1537-44, 1997. [PUBMED Abstract]
Zimmerman LE, McLean IW, Foster WD: Statistical analysis of follow-up data concerning uveal melanomas, and the influence of enucleation. Ophthalmology 87 (6): 557-64, 1980. [PUBMED Abstract]
De Potter P, Shields CL, Shields JA: New treatment modalities for uveal melanoma. Curr Opin Ophthalmol 7 (3): 27-32, 1996. [PUBMED Abstract]
The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma II: initial mortality findings. COMS report no. 10. Am J Ophthalmol 125 (6): 779-96, 1998. [PUBMED Abstract]
Hawkins BS; Collaborative Ocular Melanoma Study Group: The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma: IV. Ten-year mortality findings and prognostic factors. COMS report number 24. Am J Ophthalmol 138 (6): 936-51, 2004. [PUBMED Abstract]
Damato B: The role of eyewall resection in uveal melanoma management. Int Ophthalmol Clin 46 (1): 81-93, 2006. [PUBMED Abstract]
Bechrakis NE, Bornfeld N, Zöller I, et al.: Iodine 125 plaque brachytherapy versus transscleral tumor resection in the treatment of large uveal melanomas. Ophthalmology 109 (10): 1855-61, 2002. [PUBMED Abstract]
Bechrakis NE, Petousis V, Willerding G, et al.: Ten-year results of transscleral resection of large uveal melanomas: local tumour control and metastatic rate. Br J Ophthalmol 94 (4): 460-6, 2010. [PUBMED Abstract]
Hsueh EC, Essner R, Foshag LJ, et al.: Prolonged survival after complete resection of metastases from intraocular melanoma. Cancer 100 (1): 122-9, 2004. [PUBMED Abstract]
Pawlik TM, Zorzi D, Abdalla EK, et al.: Hepatic resection for metastatic melanoma: distinct patterns of recurrence and prognosis for ocular versus cutaneous disease. Ann Surg Oncol 13 (5): 712-20, 2006. [PUBMED Abstract]
Albert DM, Kulkarni AD: Intraocular melanoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 2090-8.
Shields CL, Shields JA, Cater J, et al.: Plaque radiotherapy for uveal melanoma: long-term visual outcome in 1106 consecutive patients. Arch Ophthalmol 118 (9): 1219-28, 2000. [PUBMED Abstract]
Diener-West M, Earle JD, Fine SL, et al.: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, III: initial mortality findings. COMS Report No. 18. Arch Ophthalmol 119 (7): 969-82, 2001. [PUBMED Abstract]
Collaborative Ocular Melanoma Study Group: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report No. 28. Arch Ophthalmol 124 (12): 1684-93, 2006. [PUBMED Abstract]
Melia M, Moy CS, Reynolds SM, et al.: Quality of life after iodine 125 brachytherapy vs enucleation for choroidal melanoma: 5-year results from the Collaborative Ocular Melanoma Study: COMS QOLS Report No. 3. Arch Ophthalmol 124 (2): 226-38, 2006. [PUBMED Abstract]
Char DH, Kroll S, Phillips TL, et al.: Late radiation failures after iodine 125 brachytherapy for uveal melanoma compared with charged-particle (proton or helium ion) therapy. Ophthalmology 109 (10): 1850-4, 2002. [PUBMED Abstract]
Gragoudas E, Li W, Goitein M, et al.: Evidence-based estimates of outcome in patients irradiated for intraocular melanoma. Arch Ophthalmol 120 (12): 1665-71, 2002. [PUBMED Abstract]
Caujolle JP, Mammar H, Chamorey E, et al.: Proton beam radiotherapy for uveal melanomas at nice teaching hospital: 16 years’ experience. Int J Radiat Oncol Biol Phys 78 (1): 98-103, 2010. [PUBMED Abstract]
Tsuji H, Ishikawa H, Yanagi T, et al.: Carbon-ion radiotherapy for locally advanced or unfavorably located choroidal melanoma: a Phase I/II dose-escalation study. Int J Radiat Oncol Biol Phys 67 (3): 857-62, 2007. [PUBMED Abstract]
Char DH, Quivey JM, Castro JR, et al.: Helium ions versus iodine 125 brachytherapy in the management of uveal melanoma. A prospective, randomized, dynamically balanced trial. Ophthalmology 100 (10): 1547-54, 1993. [PUBMED Abstract]
Gragoudas ES, Lane AM, Regan S, et al.: A randomized controlled trial of varying radiation doses in the treatment of choroidal melanoma. Arch Ophthalmol 118 (6): 773-8, 2000. [PUBMED Abstract]
Modorati G, Miserocchi E, Galli L, et al.: Gamma knife radiosurgery for uveal melanoma: 12 years of experience. Br J Ophthalmol 93 (1): 40-4, 2009. [PUBMED Abstract]
Muller K, Nowak PJ, de Pan C, et al.: Effectiveness of fractionated stereotactic radiotherapy for uveal melanoma. Int J Radiat Oncol Biol Phys 63 (1): 116-22, 2005. [PUBMED Abstract]
Dieckmann K, Georg D, Bogner J, et al.: Optimizing LINAC-based stereotactic radiotherapy of uveal melanomas: 7 years’ clinical experience. Int J Radiat Oncol Biol Phys 66 (4 Suppl 1): 47-52, 2006.
Harbour JW, Meredith TA, Thompson PA, et al.: Transpupillary thermotherapy versus plaque radiotherapy for suspected choroidal melanomas. Ophthalmology 110 (11): 2207-14; discussion 2215, 2003. [PUBMED Abstract]
Pilotto E, Vujosevic S, De Belvis V, et al.: Long-term choroidal vascular changes after iodine brachytherapy versus transpupillary thermotherapy for choroidal melanoma. Eur J Ophthalmol 19 (4): 646-53, 2009 Jul-Aug. [PUBMED Abstract]
Desjardins L, Lumbroso-Le Rouic L, Levy-Gabriel C, et al.: Combined proton beam radiotherapy and transpupillary thermotherapy for large uveal melanomas: a randomized study of 151 patients. Ophthalmic Res 38 (5): 255-60, 2006. [PUBMED Abstract]
Treatment of Iris Melanoma
Melanocytic stromal proliferations and nevi of the iris are the most common tumors of the iris, but melanoma is rare.[1,2] Clinical differentiation between an iris nevus and a melanoma might sometimes be difficult and, at times, impossible. Melanomas of the iris are usually small discrete lesions, although they may occasionally be diffuse, infiltrative, or multiple and may result in heterochromia, chronic uveitis, or spontaneous hemorrhage into the anterior chamber of the eye (hyphema). Iris melanomas that involve more than 66% of the angle circumference are associated with secondary glaucoma.[3]
Routine evaluation of iris melanomas includes gonioscopy, transillumination of the globe, and indirect ophthalmoscopy with 360° of scleral depression. Photographic documentation is essential to verify progression in size or growth of the tumor.[4] Anterior segment fluorescein angiography may be helpful to demonstrate the vascularity of the lesion but is not diagnostic. High-resolution ultrasound biomicroscopy can be used to measure small lesions (basal dimensions and thickness) and to assess tumor involvement of the anterior ciliary body, angle, and overlying sclera.[5] The main disadvantage with this technology is its limited penetration of large lesions. In these cases, conventional ultrasonography is more accurate.
In general, patients with iris melanomas have relatively good outcomes. Only about 3% of these melanomas metastasize within 5 years.[1] Iris melanomas are predominantly of the spindle-cell type and are usually smaller in size than posterior melanomas. Clinical features, including prominent tumor vascularity, rapid growth, and heterogeneous pigmentation, are associated with an epithelioid cell component.[6] Involvement of the iridocorneal angles is frequently associated with ciliary body invasion.[6]
Because iris melanomas are rare and have a good prognosis, clinical trials with sufficient power are impractical. Therefore, treatment experience is based principally on case series and case reports. Conservative management is generally advocated whenever possible, but surgical intervention may be justified with unequivocal tumor growth or extensive disease at initial examination.
Treatment Options for Iris Melanoma
Treatment options for iris melanoma include:
Observation with careful follow-up. This option is used in asymptomatic patients with stable lesions; follow-up includes serial photography.[3]
Local resection. This option is used when progressive and pronounced growth is documented.[2]
Enucleation. This option is used if the tumor is not amenable to local resection because of diffuse involvement of the iris, involvement of more than 50% of the iris and anterior chamber angle, intractable glaucoma, or extraocular extension.[7]
Plaque radiation therapy. This option is offered as an alternative for large, diffuse, surgically nonresectable lesions of the iris.[8]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Albert DM, Kulkarni AD: Intraocular melanoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 2090-8.
Starr OD, Patel DV, Allen JP, et al.: Iris melanoma: pathology, prognosis and surgical intervention. Clin Experiment Ophthalmol 32 (3): 294-6, 2004. [PUBMED Abstract]
Marcus DM, Sahel JA, Jakobiec FA, et al.: Pigmented tumors of the iris. In: Albert DM, Jakobiec FA, eds.: Principles and Practice of Ophthalmology. WB Saunders Co., 1994, pp 3198-3208.
Conway RM, Chua WC, Qureshi C, et al.: Primary iris melanoma: diagnostic features and outcome of conservative surgical treatment. Br J Ophthalmol 85 (7): 848-54, 2001. [PUBMED Abstract]
Melanocytic tumors of the iris stroma. In: Shields JA: Diagnosis and Management of Intraocular Tumors. C.V. Mosby Company, 1983, pp 83-121.
Shields CL, Shields JA, De Potter P, et al.: Treatment of non-resectable malignant iris tumours with custom designed plaque radiotherapy. Br J Ophthalmol 79 (4): 306-12, 1995. [PUBMED Abstract]
Treatment of Ciliary Body Melanoma
Melanoma involving the ciliary body is rare and patients have a poor prognosis. In some cases, diagnosis may be difficult because of similarity to other eye diseases. The differential diagnosis of ciliary body melanoma is considered in cases of unilateral pigmentary glaucoma and chronic uveitis.[1]
Ultrasound biomicroscopy can be used to evaluate tumor shape, thickness, margins, reflectivity, and local invasion.[2,3] Patients with tumors greater than 7 mm in thickness are at increased risk of metastatic disease and melanoma-related death compared with patients with thinner tumors.[4]
Treatment Options for Ciliary Body Melanoma
There are several options for management of ciliary body melanoma. All of them are reported from case series.[Level of evidence C3] The choice of therapy, however, depends on many factors.
Treatment options for ciliary body melanoma include:
Plaque radiation therapy. Local control rates are high, but treatment is associated with a high incidence of secondary cataract.[4,5]
External-beam, charged-particle radiation therapy. This approach is offered at specialized referral centers. It requires careful patient cooperation, with voluntary fixation of gaze.[6–8]
Local tumor resection. This option is mainly suitable for selected ciliary body or anterior choroidal tumors with smaller basal dimension and greater thickness.[9,10]
Enucleation. This option is generally reserved for large melanomas when regaining useful vision is unlikely. It is also indicated in the presence of intractable secondary glaucoma and extraocular extension.[5,8]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Nguyen QD, Foster CS: Ciliary body melanoma masquerading as chronic uveitis. Ocul Immunol Inflamm 6 (4): 253-6, 1998. [PUBMED Abstract]
Marigo FA, Finger PT, McCormick SA, et al.: Iris and ciliary body melanomas: ultrasound biomicroscopy with histopathologic correlation. Arch Ophthalmol 118 (11): 1515-21, 2000. [PUBMED Abstract]
Daftari I, Barash D, Lin S, et al.: Use of high-frequency ultrasound imaging to improve delineation of anterior uveal melanoma for proton irradiation. Phys Med Biol 46 (2): 579-90, 2001. [PUBMED Abstract]
Gündüz K, Shields CL, Shields JA, et al.: Plaque radiotherapy of uveal melanoma with predominant ciliary body involvement. Arch Ophthalmol 117 (2): 170-7, 1999. [PUBMED Abstract]
Finger PT: Plaque radiation therapy for malignant melanoma of the iris and ciliary body. Am J Ophthalmol 132 (3): 328-35, 2001. [PUBMED Abstract]
Munzenrider JE: Uveal melanomas. Conservation treatment. Hematol Oncol Clin North Am 15 (2): 389-402, 2001. [PUBMED Abstract]
Char DH, Kroll SM, Castro J: Ten-year follow-up of helium ion therapy for uveal melanoma. Am J Ophthalmol 125 (1): 81-9, 1998. [PUBMED Abstract]
De Potter P: [Choroidal melanoma: current therapeutic approaches] J Fr Ophtalmol 25 (2): 203-11, 2002. [PUBMED Abstract]
De Potter P, Shields CL, Shields JA: New treatment modalities for uveal melanoma. Curr Opin Ophthalmol 7 (3): 27-32, 1996. [PUBMED Abstract]
A wide range of 5-year mortality rates has been reported for patients with small choroidal melanomas, with an average rate of about 16%.[1,2] Several studies indicate that the two most important clinical factors predictive of mortality are larger tumor size at the time of treatment and documentation of tumor growth.[3]
The management of small choroidal melanomas is controversial. The likelihood of progression from the time of diagnosis to growth warranting treatment has not been well characterized. Many ophthalmologists advocate initial observation. This initial management strategy is justified on several grounds, including difficulty in establishing a correct diagnosis, lack of documented efficacy for globe-conserving treatments, and concerns for severe treatment-related morbidity. Others have advocated earlier therapeutic intervention.[4–6]
Treatment Options for Small Choroidal Melanoma
Treatment options for small choroidal melanoma include:
Observation. This strategy is important for patients with an uncertain diagnosis or whose tumor growth has not been documented. It is also used for asymptomatic patients with stable lesions (particularly older or debilitated patients) and for patients with a tumor in their only useful eye.[2]
Plaque radiation therapy. This treatment is used for small- or medium-sized uveal melanomas, amelanotic tumors, or tumors that touch the optic disc for greater than 3 clock-hours of optic disk circumference.[7,8]
External-beam, charged-particle radiation therapy. This approach is offered at specialized referral centers. It requires careful patient cooperation, with voluntary fixation of gaze.[7–10]
Gamma-knife radiation surgery. This treatment may be a feasible option for small- to medium-sized melanomas.[11–13]
Transpupillary thermotherapy. As noted above, this approach has very limited use, but it can be used as a primary treatment or as an adjunctive method to plaque radiation therapy.[5,6,14–18] For more information, see the Transpupillary Thermotherapy section.
Local tumor resection. This strategy is used mainly for selected ciliary body or anterior choroidal tumors with smaller basal dimensions and greater thickness.[19]
Enucleation. This approach is used when severe intraocular pressure elevation is a factor. It may also be considered with small- and medium-sized melanomas that are invading the tissues of the optic nerve.[20]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Diener-West M, Hawkins BS, Markowitz JA, et al.: A review of mortality from choroidal melanoma. II. A meta-analysis of 5-year mortality rates following enucleation, 1966 through 1988. Arch Ophthalmol 110 (2): 245-50, 1992. [PUBMED Abstract]
Mortality in patients with small choroidal melanoma. COMS report no. 4. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 115 (7): 886-93, 1997. [PUBMED Abstract]
Shields CL, Shields JA, Kiratli H, et al.: Risk factors for growth and metastasis of small choroidal melanocytic lesions. Ophthalmology 102 (9): 1351-61, 1995. [PUBMED Abstract]
Shields CL, Cater J, Shields JA, et al.: Combination of clinical factors predictive of growth of small choroidal melanocytic tumors. Arch Ophthalmol 118 (3): 360-4, 2000. [PUBMED Abstract]
Shields CL, Shields JA, Perez N, et al.: Primary transpupillary thermotherapy for small choroidal melanoma in 256 consecutive cases: outcomes and limitations. Ophthalmology 109 (2): 225-34, 2002. [PUBMED Abstract]
Robertson DM, Buettner H, Bennett SR: Transpupillary thermotherapy as primary treatment for small choroidal melanomas. Arch Ophthalmol 117 (11): 1512-9, 1999. [PUBMED Abstract]
Munzenrider JE: Uveal melanomas. Conservation treatment. Hematol Oncol Clin North Am 15 (2): 389-402, 2001. [PUBMED Abstract]
Char DH, Kroll SM, Castro J: Ten-year follow-up of helium ion therapy for uveal melanoma. Am J Ophthalmol 125 (1): 81-9, 1998. [PUBMED Abstract]
Woodburn R, Danis R, Timmerman R, et al.: Preliminary experience in the treatment of choroidal melanoma with gamma knife radiosurgery. J Neurosurg 93 (Suppl 3): 177-9, 2000. [PUBMED Abstract]
Modorati G, Miserocchi E, Galli L, et al.: Gamma knife radiosurgery for uveal melanoma: 12 years of experience. Br J Ophthalmol 93 (1): 40-4, 2009. [PUBMED Abstract]
Muller K, Nowak PJ, de Pan C, et al.: Effectiveness of fractionated stereotactic radiotherapy for uveal melanoma. Int J Radiat Oncol Biol Phys 63 (1): 116-22, 2005. [PUBMED Abstract]
Shields JA, Shields CL: Atlas of Intraocular Tumors. Lippincott Williams & Wilkins, 1999.
Treatment of Medium and Large Choroidal Melanoma
Eye-sparing radiation therapy, either by plaque brachytherapy or external beam, is the preferred option for most patients with medium-sized choroidal melanoma. Enucleation remains the standard therapy for large choroidal melanomas and melanomas that cause severe glaucoma or invade the optic nerve.
Treatment Options for Medium and Large Choroidal Melanoma
Tumor growth pattern is a factor in the therapeutic decision. Enucleation is considered for a diffuse melanoma or extraocular extension, but radiation therapy can be employed for less extensive disease.
Treatment options for medium and large choroidal melanoma include:
External-beam, charged-particle radiation therapy. This approach is offered at specialized referral centers. It requires careful patient cooperation, with voluntary fixation of gaze.[5–7]
Combined therapy, with ablative laser coagulation or transpupillary thermotherapy to supplement plaque treatment.[10,11] For more information, see the Transpupillary Thermotherapy section.
Enucleation. This approach is considered primarily for diffuse melanomas or extraocular extension. Enucleation may be necessary when there are radiation complications or tumor recurrences.[10]
Large choroidal melanomas
Enucleation when the tumor is judged to be too large for eye-sparing approaches.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Karvat A, Duzenli C, Ma R, et al.: The treatment of choroidal melanoma with 198 Au plaque brachytherapy. Radiother Oncol 59 (2): 153-6, 2001. [PUBMED Abstract]
Tabandeh H, Chaudhry NA, Murray TG, et al.: Intraoperative echographic localization of iodine-125 episcleral plaque for brachytherapy of choroidal melanoma. Am J Ophthalmol 129 (2): 199-204, 2000. [PUBMED Abstract]
Diener-West M, Earle JD, Fine SL, et al.: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, III: initial mortality findings. COMS Report No. 18. Arch Ophthalmol 119 (7): 969-82, 2001. [PUBMED Abstract]
Melia BM, Abramson DH, Albert DM, et al.: Collaborative ocular melanoma study (COMS) randomized trial of I-125 brachytherapy for medium choroidal melanoma. I. Visual acuity after 3 years COMS report no. 16. Ophthalmology 108 (2): 348-66, 2001. [PUBMED Abstract]
Char DH, Quivey JM, Castro JR, et al.: Helium ions versus iodine 125 brachytherapy in the management of uveal melanoma. A prospective, randomized, dynamically balanced trial. Ophthalmology 100 (10): 1547-54, 1993. [PUBMED Abstract]
Fuss M, Loredo LN, Blacharski PA, et al.: Proton radiation therapy for medium and large choroidal melanoma: preservation of the eye and its functionality. Int J Radiat Oncol Biol Phys 49 (4): 1053-9, 2001. [PUBMED Abstract]
Char DH, Kroll SM, Castro J: Ten-year follow-up of helium ion therapy for uveal melanoma. Am J Ophthalmol 125 (1): 81-9, 1998. [PUBMED Abstract]
Peyman GA, Juarez CP, Diamond JG, et al.: Ten years experience with eye wall resection for uveal malignant melanomas. Ophthalmology 91 (12): 1720-5, 1984. [PUBMED Abstract]
Seregard S, Landau I: Transpupillary thermotherapy as an adjunct to ruthenium plaque radiotherapy for choroidal melanoma. Acta Ophthalmol Scand 79 (1): 19-22, 2001. [PUBMED Abstract]
Shields JA: The expanding role of laser photocoagulation for intraocular tumors. The 1993 H. Christian Zweng Memorial Lecture. Retina 14 (4): 310-22, 1994. [PUBMED Abstract]
Treatment of Extraocular Extension and Metastatic Intraocular Melanoma
Extrascleral extension is associated with a poor prognosis. For patients with gross tumor involvement of the orbit, treatment requires orbital exenteration. However, there is no evidence that such radical surgery will prolong survival. Most patients with localized or encapsulated extraocular extension are not exenterated. This subject is controversial.[1–5]
No effective method of systemic treatment has been identified for patients with metastatic ocular melanoma. Clinical trials are an option for these patients.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Shammas HF, Blodi FC: Prognostic factors in choroidal and ciliary body melanomas. Arch Ophthalmol 95 (1): 63-9, 1977. [PUBMED Abstract]
Pach JM, Robertson DM, Taney BS, et al.: Prognostic factors in choroidal and ciliary body melanomas with extrascleral extension. Am J Ophthalmol 101 (3): 325-31, 1986. [PUBMED Abstract]
Kersten RC, Tse DT, Anderson RL, et al.: The role of orbital exenteration in choroidal melanoma with extrascleral extension. Ophthalmology 92 (3): 436-43, 1985. [PUBMED Abstract]
Hykin PG, McCartney AC, Plowman PN, et al.: Postenucleation orbital radiotherapy for the treatment of malignant melanoma of the choroid with extrascleral extension. Br J Ophthalmol 74 (1): 36-9, 1990. [PUBMED Abstract]
Gündüz K, Shields CL, Shields JA, et al.: Plaque radiotherapy for management of ciliary body and choroidal melanoma with extraocular extension. Am J Ophthalmol 130 (1): 97-102, 2000. [PUBMED Abstract]
Treatment of Recurrent Intraocular Melanoma
The prognosis for any patient with recurring or relapsing disease is poor, regardless of cell type or stage. The question and selection of further treatment depends on many factors, including the extent of the lesion, age and health of the patient, prior treatment, site of recurrence, and individual patient considerations. Surgical resection of metastases diagnosed after initial management of ocular melanoma in single-center case series of highly selected patients has been reported. The extent to which the occasional favorable outcomes are the result of strong selection factors is not clear, so this approach cannot be considered standard.[1]
Eligible patients should consider participation in clinical trials whenever possible.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Hsueh EC, Essner R, Foshag LJ, et al.: Prolonged survival after complete resection of metastases from intraocular melanoma. Cancer 100 (1): 122-9, 2004. [PUBMED Abstract]
Latest Updates to This Summary (05/16/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of intraocular melanoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Intraocular (Uveal) Melanoma Treatment is:
Shaheer A. Khan, DO (Columbia University Irving Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Intraocular (Uveal) Melanoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/eye/hp/intraocular-melanoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389482]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
The only widely proposed screening procedure for skin cancer is visual examination of the skin, including both self-examination by the patient and clinical examination by the health care provider. Mobile phone applications that evaluate skin lesions to detect skin cancer and malignant melanoma have been launched.[1] However, the use of such applications to assess skin cancer has been problematic because of the lack of evidence of their diagnostic accuracy and because they have not been studied in large-scale screening programs.[2–4] The use of convolutional neural networks to classify images of melanoma and skin cancer is a growing area of research.[5–7]
Benefits
There is insufficient evidence that population screening for skin cancer reduces skin cancer mortality. The evidence is inadequate to determine whether visual examination of the skin in asymptomatic individuals leads to a reduction in mortality from melanomatous skin cancer. Further, in asymptomatic populations, the effect of visual skin examination on mortality from nonmelanomatous skin cancers is unknown.
Magnitude of Effect: Unknown.
Study Design: Direct evidence limited to a single ecological study.
Internal Validity: Poor.
Consistency: Not applicable.
External Validity: Poor.
Harms
Based on fair—though unquantified—evidence, visual examination of the skin in asymptomatic individuals may lead to adverse consequences. These consequences include complications of diagnostic or treatment interventions (such as poor cosmetic or functional outcomes) and the psychological effects of being labeled with a potentially fatal disease. Other harmful consequences are overdiagnosis, leading to the detection of biologically benign disease that would otherwise go undetected, and possible misdiagnosis of a benign lesion as malignant.
Magnitude of Effect: Unknown.
Study Design: Case series, ecological studies.
Internal Validity: Fair.
Consistency: Fair.
External Validity: Fair.
References
Buechi R, Faes L, Bachmann LM, et al.: Evidence assessing the diagnostic performance of medical smartphone apps: a systematic review and exploratory meta-analysis. BMJ Open 7 (12): e018280, 2017. [PUBMED Abstract]
Kassianos AP, Emery JD, Murchie P, et al.: Smartphone applications for melanoma detection by community, patient and generalist clinician users: a review. Br J Dermatol 172 (6): 1507-1518, 2015. [PUBMED Abstract]
Wolf JA, Moreau JF, Akilov O, et al.: Diagnostic inaccuracy of smartphone applications for melanoma detection. JAMA Dermatol 149 (4): 422-6, 2013. [PUBMED Abstract]
Udrea A, Mitra GD, Costea D, et al.: Accuracy of a smartphone application for triage of skin lesions based on machine learning algorithms. J Eur Acad Dermatol Venereol 34 (3): 648-655, 2020. [PUBMED Abstract]
Hekler A, Utikal JS, Enk AH, et al.: Superior skin cancer classification by the combination of human and artificial intelligence. Eur J Cancer 120: 114-121, 2019. [PUBMED Abstract]
Esteva A, Kuprel B, Novoa RA, et al.: Dermatologist-level classification of skin cancer with deep neural networks. Nature 542 (7639): 115-118, 2017. [PUBMED Abstract]
Phillips M, Marsden H, Jaffe W, et al.: Assessment of Accuracy of an Artificial Intelligence Algorithm to Detect Melanoma in Images of Skin Lesions. JAMA Netw Open 2 (10): e1913436, 2019. [PUBMED Abstract]
Incidence and Mortality of Skin Cancer
There are two main types of skin cancer:
Keratinocyte carcinoma.
Basal cell carcinoma (BCC).
Squamous cell carcinoma (SCC).
Melanoma.
BCC and SCC are the most common forms of skin cancer but have substantially better prognoses than the less common, generally more aggressive melanoma.
Keratinocyte carcinoma is the most commonly occurring cancer in the United States. Its incidence appears to be increasing in some [1] but not all [2] areas of the United States. Overall U.S. incidence rates have likely been increasing for a number of years.[3,4] At least some of this increase may be attributable to increased skin cancer awareness and resultant increasing investigation and biopsy of skin lesions. A precise estimate of the total number and incidence rate of keratinocyte carcinoma is not possible because reporting to cancer registries is not required. However, it was estimated that in 2012, 5.4 million cases of keratinocyte carcinoma were diagnosed among 3.3 million people in the United States.[5] That number exceeds all other cases of cancer estimated by the American Cancer Society for 2025, which is about 2 million.[5]
Melanoma is reportable in U.S. cancer registries, so there are more reliable estimates of incidence than for keratinocyte carcinoma. In 2025, it is estimated that 104,960 individuals in the United States will be diagnosed with invasive melanoma and 107,240 will be diagnosed with melanoma in situ. Approximately 8,430 individuals will die of melanoma in 2025. Since the early 2000s, melanoma incidence rates among individuals younger than 50 years have stabilized in women but declined by about 1% per year in men. However, among individuals aged 50 years and older in recent years, the incidence rates appeared to have stabilized in men but increased by about 3% per year in women.[5] From 2013 to 2022, melanoma mortality rates declined by about 4% per year and 3% per year in men and women, respectively.[5]
A study of skin biopsy rates in relation to melanoma incidence rates obtained from the Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute indicated that much of the observed increase in incidence between 1986 and 2001 was confined to local disease and was most likely caused by overdiagnosis as a result of increased skin biopsy rates during this period.[6] A second study that used SEER data between 2002 and 2009 reported similar findings.[7]
The incidence of melanoma also increased in children and adolescents until 2001. However, between 2001 and 2022, there was a 4.3% reduction in the yearly incidence rate of melanoma among children and adolescents in the National Childhood Cancer Registry databases.[8] During that time, the average annual incidence in this group was exceptionally low (4.5 per 1 million), which may have resulted in spurious trends.[8] Nevertheless, similar trends have been seen in Sweden.[9] In the U.S. study of pediatric melanoma, nearly one-half of the patients had local disease (22% of patients had in situ disease, and 25% of patients had superficial spreading), and nearly one-half of the patients had disease with a thickness of less than one millimeter. Given that mortality from pediatric melanoma had been fairly stable during those years,[10] it is likely that the increase in incidence could be explained, at least in part, by overdiagnosis.
References
Athas WF, Hunt WC, Key CR: Changes in nonmelanoma skin cancer incidence between 1977-1978 and 1998-1999 in Northcentral New Mexico. Cancer Epidemiol Biomarkers Prev 12 (10): 1105-8, 2003. [PUBMED Abstract]
Harris RB, Griffith K, Moon TE: Trends in the incidence of nonmelanoma skin cancers in southeastern Arizona, 1985-1996. J Am Acad Dermatol 45 (4): 528-36, 2001. [PUBMED Abstract]
Rogers HW, Weinstock MA, Harris AR, et al.: Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol 146 (3): 283-7, 2010. [PUBMED Abstract]
Leiter U, Eigentler T, Garbe C: Epidemiology of skin cancer. Adv Exp Med Biol 810: 120-40, 2014. [PUBMED Abstract]
American Cancer Society: Cancer Facts and Figures 2025. American Cancer Society, 2025. Available online. Last accessed January 16, 2025.
Welch HG, Woloshin S, Schwartz LM: Skin biopsy rates and incidence of melanoma: population based ecological study. BMJ 331 (7515): 481, 2005. [PUBMED Abstract]
Weinstock MA, Lott JP, Wang Q, et al.: Skin biopsy utilization and melanoma incidence among Medicare beneficiaries. Br J Dermatol 176 (4): 949-954, 2017. [PUBMED Abstract]
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
Austin MT, Xing Y, Hayes-Jordan AA, et al.: Melanoma incidence rises for children and adolescents: an epidemiologic review of pediatric melanoma in the United States. J Pediatr Surg 48 (11): 2207-13, 2013. [PUBMED Abstract]
Lewis KG: Trends in pediatric melanoma mortality in the United States, 1968 through 2004. Dermatol Surg 34 (2): 152-9, 2008. [PUBMED Abstract]
Risk Factors for Skin Cancer
Epidemiological evidence suggests that exposure to UV radiation and the sensitivity of an individual’s skin to UV radiation are risk factors for skin cancer, although the type of exposure (high-intensity and short-duration vs. chronic exposure) and pattern of exposure (continuous vs. intermittent) may differ among the three main types of skin cancer.[1–3] In addition, genetic predisposition and the immune system may play roles in the pathogenesis of skin cancers.[4] Organ-transplant recipients receiving immunosuppressive drugs are at elevated risk of skin cancers, particularly squamous cell carcinoma (SCC). Arsenic exposure also increases the risk of cutaneous SCC.[5,6]
The incidence of melanoma rises rapidly in White individuals after age 20 years. Fair-skinned individuals exposed to the sun are at higher risk. Individuals with certain types of pigmented lesions (dysplastic or atypical nevi), with several large nondysplastic nevi, many small nevi, or moderate freckling have a twofold to threefold increased risk of developing melanoma.[7] Individuals with familial dysplastic nevus syndrome or with several dysplastic or atypical nevi are at high (>fivefold) risk of developing melanoma.[4,7]
It is important to note that, for the general population, most melanomas may not arise from preexisting nevi. A meta-analysis of studies published between 1948 and 2016 found that the prevalence of nevus-associated melanomas was only 29%, compared with 71% for the prevalence of de novo melanomas.[8]
References
Koh HK: Cutaneous melanoma. N Engl J Med 325 (3): 171-82, 1991. [PUBMED Abstract]
Preston DS, Stern RS: Nonmelanoma cancers of the skin. N Engl J Med 327 (23): 1649-62, 1992. [PUBMED Abstract]
English DR, Armstrong BK, Kricker A, et al.: Case-control study of sun exposure and squamous cell carcinoma of the skin. Int J Cancer 77 (3): 347-53, 1998. [PUBMED Abstract]
Hawkes JE, Truong A, Meyer LJ: Genetic predisposition to melanoma. Semin Oncol 43 (5): 591-597, 2016. [PUBMED Abstract]
Thomas VD, Aasi SZ, Wilson LD, et al.: Cancer of the skin. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds.: Cancer: Principles and Practice of Oncology. Vols. 1 & 2. 8th ed. Lippincott Williams & Wilkins, 2008, pp 1863-87.
Le Mire L, Hollowood K, Gray D, et al.: Melanomas in renal transplant recipients. Br J Dermatol 154 (3): 472-7, 2006. [PUBMED Abstract]
Gandini S, Sera F, Cattaruzza MS, et al.: Meta-analysis of risk factors for cutaneous melanoma: I. Common and atypical naevi. Eur J Cancer 41 (1): 28-44, 2005. [PUBMED Abstract]
Pampena R, Kyrgidis A, Lallas A, et al.: A meta-analysis of nevus-associated melanoma: Prevalence and practical implications. J Am Acad Dermatol 77 (5): 938-945.e4, 2017. [PUBMED Abstract]
Accuracy of Making a Clinical Diagnosis of Melanoma
Observer variability among physicians has been noted in the evaluation of skin lesions and subsequent biopsy specimens. A systematic review of 32 studies that compared the accuracy of dermatologists and primary care physicians in making a clinical diagnosis of melanoma concluded that there was no statistically significant difference in accuracy. However, the results were inconclusive, owing to small sample sizes and study design weaknesses.[1] Subsequent studies have noted a higher accuracy for dermatologists in the diagnosis of melanocytic lesions,[2,3] yet there is a shortage of dermatologists to meet the demands of population-level screening.
A study of 187 pathologists who practiced in the United States found that cases of moderately dysplastic nevi to early-stage invasive melanoma had less than 50% agreement with a reference diagnosis defined by consensus of experienced pathologists.[4] At a U.S. population level, it is estimated that 82.8% (95% confidence interval, 81.0%–84.5%) of melanocytic skin biopsy diagnoses would be verified if they were reviewed by a consensus reference panel of experienced pathologists.[4] In addition, differentiating between benign and malignant melanocytic tumors during histological examinations of biopsy specimens has been shown to be inconsistent, even in the hands of experienced dermatopathologists.[5,6] This variability in the diagnosis of melanocytic lesions undermines the results of studies that examine screening effectiveness and also may undermine the effectiveness of any screening intervention. Furthermore, this finding suggests that requesting a second opinion regarding the pathology of biopsy specimens may be important.[5–7] A standardized approach to pathologists’ classifying of the interpretations of melanocytic skin lesions may also reduce confusion and improve communication between clinicians.[4,6,8,9]
References
Chen SC, Bravata DM, Weil E, et al.: A comparison of dermatologists’ and primary care physicians’ accuracy in diagnosing melanoma: a systematic review. Arch Dermatol 137 (12): 1627-34, 2001. [PUBMED Abstract]
Chen SC, Pennie ML, Kolm P, et al.: Diagnosing and managing cutaneous pigmented lesions: primary care physicians versus dermatologists. J Gen Intern Med 21 (7): 678-82, 2006. [PUBMED Abstract]
Corbo MD, Wismer J: Agreement between dermatologists and primary care practitioners in the diagnosis of malignant melanoma: review of the literature. J Cutan Med Surg 16 (5): 306-10, 2012 Sep-Oct. [PUBMED Abstract]
Elmore JG, Barnhill RL, Elder DE, et al.: Pathologists’ diagnosis of invasive melanoma and melanocytic proliferations: observer accuracy and reproducibility study. BMJ 357: j2813, 2017. [PUBMED Abstract]
Farmer ER, Gonin R, Hanna MP: Discordance in the histopathologic diagnosis of melanoma and melanocytic nevi between expert pathologists. Hum Pathol 27 (6): 528-31, 1996. [PUBMED Abstract]
Lott JP, Elmore JG, Zhao GA, et al.: Evaluation of the Melanocytic Pathology Assessment Tool and Hierarchy for Diagnosis (MPATH-Dx) classification scheme for diagnosis of cutaneous melanocytic neoplasms: Results from the International Melanoma Pathology Study Group. J Am Acad Dermatol 75 (2): 356-63, 2016. [PUBMED Abstract]
Piepkorn MW, Longton GM, Reisch LM, et al.: Assessment of Second-Opinion Strategies for Diagnoses of Cutaneous Melanocytic Lesions. JAMA Netw Open 2 (10): e1912597, 2019. [PUBMED Abstract]
Piepkorn MW, Barnhill RL, Elder DE, et al.: The MPATH-Dx reporting schema for melanocytic proliferations and melanoma. J Am Acad Dermatol 70 (1): 131-41, 2014. [PUBMED Abstract]
Radick AC, Reisch LM, Shucard HL, et al.: Terminology for melanocytic skin lesions and the MPATH-Dx classification schema: A survey of dermatopathologists. J Cutan Pathol 48 (6): 733-738, 2021. [PUBMED Abstract]
Evidence of Benefit Associated With Screening
More than 90% of melanomas that arise in the skin can be recognized with the naked eye. Very often there is a prolonged horizontal growth phase, during which the tumor expands centrifugally beneath the epidermis but does not invade the underlying dermis. This horizontal growth phase may provide lead time for early detection. Melanoma is more easily cured if treated before the onset of the vertical growth phase with its metastatic potential.[1]
The probability of tumor recurrence within 10 years after curative resection is less than 10% with tumors less than 1.4 mm in thickness. For patients with tumors less than 0.76 mm in thickness, the likelihood of recurrence is less than 1% in 10 years.[2]
A systematic review of skin cancer screening examined evidence available through mid-2005. The review concluded that direct evidence of improved health outcomes associated with skin cancer screening is lacking.[3] An updated review published in 2016 found limited evidence that skin cancer screening reduces melanoma mortality.[4,5]
No randomized trials evaluating the efficacy of skin cancer screening on mortality have been completed. A population-based trial (using cluster randomization) to determine the effect of skin cancer screening on melanoma mortality was initiated in Queensland, Australia, but lost its funding after the initial pilot phase, and no health outcomes were ever reported.[6]
Two ecological studies have been conducted using data from Germany. The first study was a pilot project conducted in 2003 and 2004, in which a skin cancer screening program was implemented in one federal state. Suggestion of a reduction in melanoma mortality with screening led to the establishment of countywide skin cancer screening programs in 2008.[7,8] The programs offered a whole-body skin examination once every 2 years for individuals older than 35 years. The second ecological study compared the melanoma mortality experience in Germany with the melanoma mortality experience of subregions of 22 European countries—none of which had organized screening programs—for the years 2000 to 2013. After adjustment for potential confounders, Germany and the 22 European regions had similar malignant mortality rates, suggesting no benefit of screening.[9]
References
Friedman RJ, Rigel DS, Kopf AW: Early detection of malignant melanoma: the role of physician examination and self-examination of the skin. CA Cancer J Clin 35 (3): 130-51, 1985 May-Jun. [PUBMED Abstract]
Blois MS, Sagebiel RW, Abarbanel RM, et al.: Malignant melanoma of the skin. I. The association of tumor depth and type, and patient sex, age, and site with survival. Cancer 52 (7): 1330-41, 1983. [PUBMED Abstract]
Wolff T, Tai E, Miller T: Screening for skin cancer: an update of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 150 (3): 194-8, 2009. [PUBMED Abstract]
Wernli KJ, Henrikson NB, Morrison CC, et al.: Screening for Skin Cancer in Adults: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 316 (4): 436-47, 2016. [PUBMED Abstract]
Bibbins-Domingo K, Grossman DC, Curry SJ, et al.: Screening for Skin Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 316 (4): 429-35, 2016. [PUBMED Abstract]
Aitken JF, Elwood JM, Lowe JB, et al.: A randomised trial of population screening for melanoma. J Med Screen 9 (1): 33-7, 2002. [PUBMED Abstract]
Katalinic A, Waldmann A, Weinstock MA, et al.: Does skin cancer screening save lives? An observational study comparing trends in melanoma mortality in regions with and without screening. Cancer 118 (21): 5395-402, 2012. [PUBMED Abstract]
Eisemann N, Waldmann A, Holleczek B, et al.: Observed and expected mortality in the German skin cancer screening pilot project SCREEN. J Med Screen 25 (3): 166-168, 2018. [PUBMED Abstract]
Kaiser M, Schiller J, Schreckenberger C: The effectiveness of a population-based skin cancer screening program: evidence from Germany. Eur J Health Econ 19 (3): 355-367, 2018. [PUBMED Abstract]
Evidence of Harms Associated With Screening
Harms have not been well studied or reported in quantitative terms, but the potential for adverse consequences from skin cancer screening exists. In the SCREEN pilot project in Germany, 4.4% of all screened participants underwent a skin excision for a suspicious lesion, but most biopsies did not result in a cancer diagnosis. The detection rate was especially affected by age. One case of melanoma was detected per 28 excisions overall (for both men and women), while 52 skin excisions were required to detect one melanoma in men aged 20 to 34 years.[1]
Visual examination of the skin in asymptomatic individuals may lead to cosmetic or functional complications of diagnostic or treatment interventions and psychological effects of being labeled with a potentially fatal disease. However, robust data on the frequency of such events are lacking. Other harmful consequences are overdiagnosis, leading to the detection of biologically benign disease that would otherwise go undetected,[2–4] and possible misdiagnosis of a benign lesion as malignant. For more information, see the Accuracy of Making a Clinical Diagnosis of Melanoma section.
References
Waldmann A, Nolte S, Geller AC, et al.: Frequency of excisions and yields of malignant skin tumors in a population-based screening intervention of 360,288 whole-body examinations. Arch Dermatol 148 (8): 903-10, 2012. [PUBMED Abstract]
Welch HG, Woloshin S, Schwartz LM: Skin biopsy rates and incidence of melanoma: population based ecological study. BMJ 331 (7515): 481, 2005. [PUBMED Abstract]
Weinstock MA, Lott JP, Wang Q, et al.: Skin biopsy utilization and melanoma incidence among Medicare beneficiaries. Br J Dermatol 176 (4): 949-954, 2017. [PUBMED Abstract]
Schoffer O, Schülein S, Arand G, et al.: Tumour stage distribution and survival of malignant melanoma in Germany 2002-2011. BMC Cancer 16 (1): 936, 2016. [PUBMED Abstract]
Latest Updates to This Summary (05/09/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Updated statistics with estimated new cases of melanoma and melanoma in situ and deaths due to melanoma for 2025. Also revised text to state that from 2013 to 2022, melanoma mortality rates declined by about 4% per year and 3% per year in men and women, respectively.
This summary is written and maintained by the PDQ Screening and Prevention Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about skin cancer screening. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Screening and Prevention Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Screening and Prevention Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Screening and Prevention Editorial Board. PDQ Skin Cancer Screening. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/skin/hp/skin-screening-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389300]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
The information in these summaries should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Melanoma is a malignant tumor of melanocytes, which are the cells that make the pigment melanin and are derived from the neural crest. Although most melanomas arise in the skin, they may also arise from mucosal surfaces or at other sites to which neural crest cells migrate, including the uveal tract. Uveal melanomas differ significantly from cutaneous melanoma in incidence, prognostic factors, molecular characteristics, and treatment. For more information, visit Intraocular (Uveal) Melanoma Treatment.
Incidence and Mortality
Estimated new cases and deaths from melanoma in the United States in 2025:[1]
New cases: 104,960.
Deaths: 8,430.
Skin cancer is the most common malignancy diagnosed in the United States. Invasive melanoma represents about 1% of skin cancers but results in the most deaths.[1,2] Since the early 2000s, the incidence of melanoma in people younger than 50 years has declined by about 1% per year in men and stabilized in women. In people aged 50 years and older, the incidence has stabilized in men and increased by about 3% per year in women.[1] Older men are at highest risk of melanoma; however, it is the most common cancer in young adults aged 25 to 29 years and the second most common cancer in those aged 15 to 29 years.[3] Ocular melanoma is the most common cancer of the eye, with approximately 2,000 cases diagnosed annually.
Risk Factors
Risk factors for melanoma include both intrinsic (genetic and phenotype) and extrinsic (environmental or exposure) factors:
EnlargeFigure 1. Schematic representation of normal skin. Melanocytes are also present in normal skin and serve as the source cell for melanoma. The relatively avascular epidermis houses both basal cell keratinocytes and squamous epithelial keratinocytes, the source cells for basal cell carcinoma and squamous cell carcinoma, respectively. The separation between epidermis and dermis occurs at the basement membrane zone, located just inferior to the basal cell keratinocytes.
Melanoma occurs predominantly in adults, and more than 50% of the cases arise in apparently normal areas of the skin. Melanoma can occur anywhere, including on mucosal surfaces and the uvea. However, in women it occurs more commonly on the extremities, and in men it occurs most commonly on the trunk or head and neck.[4]
Early signs in a nevus that would suggest a malignant change include:
Darker or variable discoloration.
Itching.
An increase in size or the development of satellite lesions.
Ulceration or bleeding (later signs).
A common acronym used by medical professionals and the lay public to identify the suspicious features of pigmented lesions that may reflect malignant change is ABCDE:[5]
Asymmetry of the lesion.
Border irregularity.
Color variation.
Diameter >6 mm.
Evolution or change in the lesion.
EnlargeFigure 2. Melanomas with characteristic asymmetry, border irregularity, color variation, and large diameter.
Diagnosis
A biopsy, preferably by local excision, should be performed for any suspicious lesions. Suspicious lesions should never be shaved off or cauterized. An experienced pathologist should examine the specimens to allow for microstaging.
Studies show that distinguishing between benign pigmented lesions and early melanomas can be difficult, and even experienced dermatopathologists can have differing opinions. To reduce the possibility of misdiagnosis for an individual patient, a second review by an independent qualified pathologist should be considered.[6,7] Agreement between pathologists in the histological diagnosis of melanomas and benign pigmented lesions has been found to be considerably variable.[6,7]
Evidence (discordance in histological evaluation):
One study found that there was discordance in the diagnosis of melanoma versus benign lesions in 37 of 140 cases examined by a panel of experienced dermatopathologists. For the histological classification of cutaneous melanoma, the highest concordance was attained for Breslow thickness and presence of ulceration, while the agreement was poor for other histological features such as Clark level of invasion, presence of regression, and lymphocytic infiltration.[6]
In another study, 38% of cases examined by a panel of expert pathologists had two or more discordant interpretations.[7]
Prognostic Factors
Prognosis is affected by the characteristics of primary and metastatic tumors. The most important prognostic factors have been incorporated into the 2017 eighth edition of the American Joint Committee on Cancer (AJCC) staging manual and include:[4,8–10]
Thickness and/or level of invasion of the melanoma.
Ulceration or bleeding at the primary site.
Number of regional lymph nodes involved, with distinction of clinically occult and clinically apparent.
Presence of non-nodal regional disease, including microsatellites, satellites, and in-transit cutaneous or subcutaneous metastases.
Systemic metastasis.
Site—nonvisceral versus lung versus all other visceral sites versus central nervous system.
Elevated serum lactate dehydrogenase level.
Patients who are younger, female, and who have melanomas on their extremities generally have better prognoses.[4,8,9,11]
The risk of relapse decreases substantially over time, although late relapses do occur.[12–15] Long-term follow-up is important for detection of recurrence, managing long-term effects, and surveillance of new lesions.
Related Subtypes
Mucosal melanoma arises from melanocytes within the mucosal lining of the respiratory, gastrointestinal, or genitourinary tracts. This is a rare subgroup, representing only 1.4% of melanomas.[16] The etiology of mucosal melanomas remains unclear, but whole-genome sequencing reveals mutational signatures unrelated to UV radiation, which is distinct from cutaneous melanomas.[17] Diagnosis is often delayed due to the location of these lesions, a lack of symptoms, and potential amelanotic appearance.[18] There is no clear consensus on staging definitions for mucosal melanoma, and the AJCC eighth edition TNM (tumor, node, metastasis) staging is only used for head and neck mucosal melanomas.[10] The overall prognosis is poor and varies by location.
References
American Cancer Society: Cancer Facts and Figures 2025. American Cancer Society, 2025. Available online. Last accessed January 16, 2025.
Melanoma. Bethesda, Md: National Library of Medicine, 2012. Available online. Last accessed May 1, 2025.
Bleyer A, O’Leary M, Barr R, et al., eds.: Cancer Epidemiology in Older Adolescents and Young Adults 15 to 29 Years of Age, Including SEER Incidence and Survival: 1975-2000. National Cancer Institute, 2006. NIH Pub. No. 06-5767. Available online. Last accessed May 1, 2025.
Slingluff CI Jr, Flaherty K, Rosenberg SA, et al.: Cutaneous melanoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 1643-91.
Abbasi NR, Shaw HM, Rigel DS, et al.: Early diagnosis of cutaneous melanoma: revisiting the ABCD criteria. JAMA 292 (22): 2771-6, 2004. [PUBMED Abstract]
Corona R, Mele A, Amini M, et al.: Interobserver variability on the histopathologic diagnosis of cutaneous melanoma and other pigmented skin lesions. J Clin Oncol 14 (4): 1218-23, 1996. [PUBMED Abstract]
Farmer ER, Gonin R, Hanna MP: Discordance in the histopathologic diagnosis of melanoma and melanocytic nevi between expert pathologists. Hum Pathol 27 (6): 528-31, 1996. [PUBMED Abstract]
Balch CM, Soong S, Ross MI, et al.: Long-term results of a multi-institutional randomized trial comparing prognostic factors and surgical results for intermediate thickness melanomas (1.0 to 4.0 mm). Intergroup Melanoma Surgical Trial. Ann Surg Oncol 7 (2): 87-97, 2000. [PUBMED Abstract]
Manola J, Atkins M, Ibrahim J, et al.: Prognostic factors in metastatic melanoma: a pooled analysis of Eastern Cooperative Oncology Group trials. J Clin Oncol 18 (22): 3782-93, 2000. [PUBMED Abstract]
Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 563–85.
Balch CM, Gershenwald JE, Soong SJ, et al.: Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 27 (36): 6199-206, 2009. [PUBMED Abstract]
Shen P, Guenther JM, Wanek LA, et al.: Can elective lymph node dissection decrease the frequency and mortality rate of late melanoma recurrences? Ann Surg Oncol 7 (2): 114-9, 2000. [PUBMED Abstract]
Tsao H, Cosimi AB, Sober AJ: Ultra-late recurrence (15 years or longer) of cutaneous melanoma. Cancer 79 (12): 2361-70, 1997. [PUBMED Abstract]
Sarac E, Wilhelmi J, Thomas I, et al.: Late recurrence of melanoma after 10 years – Is the course of the disease different from early recurrences? J Eur Acad Dermatol Venereol 34 (5): 977-983, 2020. [PUBMED Abstract]
Faries MB, Steen S, Ye X, et al.: Late recurrence in melanoma: clinical implications of lost dormancy. J Am Coll Surg 217 (1): 27-34; discussion 34-6, 2013. [PUBMED Abstract]
McLaughlin CC, Wu XC, Jemal A, et al.: Incidence of noncutaneous melanomas in the U.S. Cancer 103 (5): 1000-7, 2005. [PUBMED Abstract]
Hayward NK, Wilmott JS, Waddell N, et al.: Whole-genome landscapes of major melanoma subtypes. Nature 545 (7653): 175-180, 2017. [PUBMED Abstract]
Thomas NE, Kricker A, Waxweiler WT, et al.: Comparison of clinicopathologic features and survival of histopathologically amelanotic and pigmented melanomas: a population-based study. JAMA Dermatol 150 (12): 1306-314, 2014. [PUBMED Abstract]
Cellular and Molecular Classification of Melanoma
The descriptive terms for clinicopathological cellular subtypes of malignant melanoma are of historical interest only; they do not have independent prognostic or therapeutic significance. These cellular subtypes include:
Superficial spreading.
Nodular.
Lentigo maligna.
Acral lentiginous (palmar/plantar and subungual).
Miscellaneous unusual types:
Mucosal lentiginous (oral and genital).
Desmoplastic.
Verrucous.
Genomic Classification
Cutaneous melanoma
The Cancer Genome Atlas (TCGA) Network performed an integrative multiplatform characterization of 333 cutaneous melanomas from 331 patients.[1] Using six types of molecular analysis at the DNA, RNA, and protein levels, the researchers identified four major genomic subtypes:
BRAF-altered.
RAS-altered.
NF1-altered.
Triple wild-type.
Genomic subtypes may suggest drug targets and clinical trial design, as well as guide clinical decision-making for targeted therapies. For more information, visit Table 1.
Targeted therapies have demonstrated efficacy and received U.S. Food and Drug Administration approval for the BRAF-altered subtype of melanoma. Combination therapies with a BRAF plus a MEK inhibitor have shown improvement in outcomes over a single-agent inhibitor alone. However, virtually all patients acquire resistance to this therapy and experience disease relapse. Therefore, clinical trials remain an important option for patients with melanoma and a BRAF pathogenic variant and other genomic subtypes of melanoma. For more information, visit the individual treatment sections.
A variety of immunotherapies have been approved for the treatment of melanoma regardless of genetic subtype. The benefit of immunotherapy has not been associated with a specific pathogenic variant or molecular subtype. The TCGA analysis identified immune markers (in a subset within each molecular subtype) that were associated with improved survival and that may have implications for immunotherapy. Identification of predictive biomarkers remains an active area of research. For more information, visit the individual treatment sections.
Table 1. Multiplatform Analysis: Pathogenic Variant, Copy Number, Whole Genome, miRNA/RNA Expression, Protein Expression in Cutaneous Melanomaa
Genomic Subtype
% Samples With Pathogenic Variant
Increased Lymphocytic Infiltration (%)
Clinical Management Implications for Targeted Therapyb,c
FDA = U.S. Food and Drug Administration; WT = wild-type.
aPrimary melanoma with matched normal samples; N = 67 (20%). Metastatic melanoma with matched normal samples; N = 266 (80%). Matched is defined as sample from the same patient.
bThe indications for immunotherapy are not known to be determined or limited by genomic subtype.
Uveal melanomas differ significantly from cutaneous melanomas. ln one series, 83% of 186 uveal melanomas were found to have a constitutively active somatic pathogenic variant in GNAQ or GNA11.[2,3] For more information, visit Intraocular (Uveal) Melanoma Treatment.
References
Cancer Genome Atlas Network: Genomic Classification of Cutaneous Melanoma. Cell 161 (7): 1681-96, 2015. [PUBMED Abstract]
Van Raamsdonk CD, Bezrookove V, Green G, et al.: Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457 (7229): 599-602, 2009. [PUBMED Abstract]
Van Raamsdonk CD, Griewank KG, Crosby MB, et al.: Mutations in GNA11 in uveal melanoma. N Engl J Med 363 (23): 2191-9, 2010. [PUBMED Abstract]
Stage Information for Melanoma
Clinical staging is based on the thickness and ulceration status of the primary tumor, and whether the tumor has spread to regional lymph nodes or distant sites. For melanoma that is clinically confined to the primary site, the chance of lymph node or systemic metastases increases as the thickness and depth of local invasion increases, which worsens the prognosis. Melanoma can spread by local extension (through lymphatics) and/or by hematogenous routes to distant sites. Any organ may be involved by metastases, but the lungs and liver are common sites.
The microstage of malignant melanoma is determined on histological examination by the vertical thickness of the lesion in millimeters (Breslow classification) and/or the anatomical level of local invasion (Clark classification). The Breslow thickness is more reproducible and more accurately predicts subsequent behavior of malignant melanoma in lesions thicker than 1.5 mm and should always be reported.
Accurate microstaging of the primary tumor requires careful histological evaluation of the entire specimen by an experienced pathologist.
Clark Classification (Level of Invasion)
Table 2. Clark Classification (Level of Invasion)
Level of Invasion
Description
Level I
Lesions involving only the epidermis (in situ melanoma); not an invasive lesion.
Level II
Invasion of the papillary dermis; does not reach the papillary-reticular dermal interface.
Level III
Invasion fills and expands the papillary dermis but does not penetrate the reticular dermis.
Level IV
Invasion into the reticular dermis but not into the subcutaneous tissue.
Level V
Invasion through the reticular dermis into the subcutaneous tissue.
AJCC Stage Groupings and TNM Definitions
The American Joint Committee on Cancer (AJCC) has designated staging by TNM (tumor, node, metastasis) classification to define melanoma.[1]
Cancers staged using this staging system include cutaneous melanoma. Cancers not staged using this system include melanoma of the conjunctiva; melanoma of the uvea; mucosal melanoma arising in the head and neck; mucosal melanoma of the urethra, vagina, rectum, and anus; Merkel cell carcinoma; and squamous cell carcinoma.[1]
AJCC Prognostic Stage Groups-Clinical (cTNM)
Table 3. Definition of cTNM Stage 0a
Stage
TNM
T Category (Thickness/Ulceration Status)
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH Level)
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical; LDH = lactate dehydrogenase; No. = number.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
bThickness and ulceration status not applicable.
0
Tis, N0, M0
Tis = Melanoma in situ.b
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
Table 4. Definition of cTNM Stages IA and IBa
Stage
TNM
T Category (Thickness/Ulceration Status)
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH Level)
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical; LDH = lactate dehydrogenase; No. = number.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
IA
T1a, N0, M0
T1a = <0.8 mm/without ulceration.
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
IB
T1b, N0, M0
T1b = <0.8 mm with ulceration; 0.8–1.0 mm with or without ulceration.
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
T2a, N0, M0
T2a = >1.0–2.0 mm/without ulceration.
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
Table 5. Definition of cTNM Stages IIA, IIB, and IICa
Stage
TNM
T Category (Thickness/Ulceration Status)
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH Level)
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical; LDH = lactate dehydrogenase; No. = number.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
IIA
T2b, N0, M0
T2b = >1.0–2.0 mm/with ulceration.
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
T3a, N0, M0
T3a = >2.0–4.0 mm/without ulceration.
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
IIB
T3b, N0, M0
T3b = >2.0–4.0 mm/with ulceration.
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
T4a, N0, M0
T4a = >4.0 mm/without ulceration.
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
IIC
T4b, N0, M0
T4b = >4.0 mm/with ulceration.
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
Table 6. Definition of cTNM Stage IIIa
Stage
TNM
T Category (Thickness/Ulceration Status)
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH)
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical; LDH = lactate dehydrogenase; No. = number.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
bFor example, diagnosis by curettage.
cFor example, unknown primary or completely regressed melanoma.
dThickness and ulceration status not applicable.
eDetected by sentinel lymph node biopsy.
III
Any T, Tis, ≥N1, M0
TX = Primary tumor cannot be assessed.b,d
N1a = One clinically occult nodee /in-transit, satellite, and/or microsatellite metastases not present.
M0 = No evidence of distant metastasis.
T0 = No evidence of primary tumor.c,d
Tis = Melanoma in situ.d
N1b = One clinically detected node/in-transit, satellite, and/or microsatellite metastases not present.
T1b = <0.8 mm with ulceration; 0.8–1.0 mm with or without ulceration.
N2a = Two or three clinically occult nodese/in-transit, satellite, and/or microsatellite metastases not present.
T2a = >1.0–2.0 mm/without ulceration.
N2b = Two or three nodes at least one of which was clinically detected/in-transit, satellite, and or microsatellite metastases not present.
T2b = >1.0–2.0 mm/with ulceration.
N2c = One clinically occult or clinically detected node/in-transit, satellite, and/or microsatellite metastases present.
T3a = >2.0–4.0 mm/without ulceration.
N3a = Four or more clinically occult nodese/in-transit, satellite, and/or microsatellite metastases not present.
T3b = >2.0–4.0 mm/with ulceration.
N3b = Four or more nodes, at least one of which was clinically detected, or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases not present.
T4a = >4.0 mm/without ulceration.
N3c = Two or more clinically occult or clinically detected nodes and/or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases present.
T4b = >4.0 mm/with ulceration.
Table 7. Definition of cTNM Stage IVa
Stage
TNM
T Category (Thickness/Ulceration Status)
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH Level)
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical; CNS = central nervous system; LDH = lactate dehydrogenase; No. = number.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
bFor example, sentinel lymph node biopsy not performed, regional nodes previously removed for another reason. (Exception: pathological N category is not required for T1 melanomas, use cN).
NX = Regional nodes not assessed;b N0 = No regional metastases; ≥N1 = Visit Table 6 for description.
M1 = Evidence of distant metastasis.
–M1a = Distant metastasis to skin, soft tissue including muscle, and/or nonregional lymph nodes [M1a(0) = LDH not elevated; M1a(1) = LDH elevated].
–M1b = Distant metastasis to lung with or without M1a sites of disease [M1b(0) = LDH not elevated; M1b(1) = LDH elevated].
–M1c = Distant metastasis to non-CNS visceral sites with or without M1a or M1b sites of disease [M1c(0) = LDH not elevated; OR M1c(1) = LDH elevated].
–M1d = Distant metastasis to CNS with or without M1a, M1b, or M1c sites of disease [M1d(0) = LDH not elevated; M1d(1) = LDH elevated].
AJCC Prognostic Stage Groups-Pathological (pTNM)
Table 8. Definition of pTNM Stage 0a,b
Stage
TNM
T Category (Thickness/Ulceration Status)
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH Level)
Illustration
T = primary tumor; N = regional lymph node; M = distant metastasis; cN = clinical N; LDH = lactate dehydrogenase; No. = number; p = pathological.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
bPathological stage 0 (melanoma in situ) and T1 do not require pathological evaluation of lymph nodes to complete pathological staging; use cN information to assign their pathological stage.
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH Level)
Illustration
T = primary tumor; N = regional lymph node; M = distant metastasis; cN = clinical N; LDH = lactate dehydrogenase; No. = number; p = pathological.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
bPathological stage 0 (melanoma in situ) and T1 do not require pathological evaluation of lymph nodes to complete pathological staging; use cN information to assign their pathological stage.
T1b = <0.8 mm with ulceration; 0.8–1.0 mm with or without ulceration.
IB
T2a, N0, M0
T2a = >1.0–2.0 mm/without ulceration.
N0 = No regional metastases detected.
M0 = No evidence of distant metastasis.
Table 10. Definition of pTNM Stages IIA, IIB, and IICa
Stage
TNM
T Category (Thickness/Ulceration Status)
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH Level)
Illustration
T = primary tumor; N = regional lymph node; M = distant metastasis; LDH = lactate dehydrogenase; No. = number; p = pathological.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
Table 11. Definition of pTNM Stages IIIA, IIIB, IIIC, and IIIDa
Stage
TNM
T Category (Thickness/Ulceration Status)
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH Level)
T = primary tumor; N = regional lymph node; M = distant metastasis; LDH = lactate dehydrogenase; No. = number; p = pathological.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
bDetected by sentinel lymph node biopsy.
cFor example, unknown primary or completely regressed melanoma.
dThickness and ulceration status not applicable.
IIIA
T1a/b–T2a, N1a or N2a, M0
T1a = <0.8 mm/without ulceration/T1b = <0.8 mm with ulceration; 0.8–1.0 mm with or without ulceration.
N1a = One clinically occult nodeb/in-transit, satellite, and/or microsatellite metastases not present; OR N2a = Two or three clinically occult nodesb/in-transit, satellite, and/or microsatellite metastases not present.
M0 = No evidence of distant metastasis.
T2a = >1.0–2.0 mm/without ulceration.
IIIB
T0, N1b, N1c, M0
T0 = No evidence of primary tumor.c,d
N1b = One clinically detected node/in-transit, satellite, and/or microsatellite metastases not present.
N2a = Two or three clinically occult nodesb/in-transit, satellite, and/or microsatellite metastases not present.
N2b = Two or three nodes, at least one of which was clinically detected/in-transit, satellite, and/or microsatellite metastases not present.
IIIC
T0, N2b, N2c, N3b, or N3c, M0
T0 = No evidence of primary tumor.c,d
N2b = Two or three nodes, at least one of which was clinically detected/in-transit, satellite, and/or microsatellite metastases not present.
M0 = No evidence of distant metastasis.
N2c = One clinically occult or clinically detected node/in-transit, satellite, and/or microsatellite metastases present.
N3b = Four or more nodes, at least one of which was clinically detected, or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases not present; OR
N3c = Two or more clinically occult or clinically detected nodes and/or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases present.
T1a–T3a, N2c or N3a/b/c, M0
T1a = <0.8 mm/without ulceration/T1b = <0.8 mm with ulceration; 0.8–1.0 mm with or without ulceration.
N2c = One clinically occult or clinically detected node/in-transit, satellite, and/or microsatellite metastases present; OR
M0 = No evidence of distant metastasis.
T2a = >1.0–2.0 mm/without ulceration.
T2b = >1.0–2.0 mm/with ulceration.
N3a = Four or more clinically occult nodesb/in-transit, satellite, and/or microsatellite metastases not present.
N3b = Four or more nodes, at least one of which was clinically detected, or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases not present.
T3a = >2.0–4.0 mm/without ulceration.
N3c = Two or more clinically occult or clinically detected nodes and/or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases present.
N2a = Two or three clinically occult nodesb/in-transit, satellite, and/or microsatellite metastases not present.
N2b = Two or three nodes, at least one of which was clinically detected/in-transit, satellite, and/or microsatellite metastases not present.
N2c = One clinically occult or clinically detected node/in-transit, satellite, and/or microsatellite metastases present.
N3a = Four or more clinically occult nodesb/in-transit, satellite, and/or microsatellite metastases not present.
N3b = Four or more nodes, at least one of which was clinically detected, or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases not present.
N3c = Two or more clinically occult or clinically detected nodes and/or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases present.
T4b, N1a–N2c, M0
T4b = >4.0 mm/with ulceration.
N1a = One clinically occult nodeb/in-transit, satellite, and/or microsatellite metastases not present.
M0 = No evidence of distant metastasis.
N1b = One clinically detected node/in-transit, satellite, and/or microsatellite metastases not present.
N2a = Two or three clinically occult nodesb/in-transit, satellite, and/or microsatellite metastases not present.
N2b = Two or three nodes, at least one of which was clinically detected/in-transit, satellite, and/or microsatellite metastases not present.
N2c = One clinically occult or clinically detected node/in-transit, satellite, and/or microsatellite metastases present.
IIID
T4b, N3a/b/c, M0
T4b = >4.0 mm/with ulceration.
N3a = Four or more clinically occult nodesb/in-transit, satellite, and/or microsatellite metastases not present.
M0 = No evidence of distant metastasis.
N3b = Four or more nodes, at least one of which was clinically detected, or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases not present.
N3c = Two or more clinically occult or clinically detected nodes and/or presence of any number of matted nodes/in-transit, satellite, and/or microsatellite metastases present.
Table 12. Definition of pTNM Stage IVa
Stage
TNM
T Category (Thickness/Ulceration Status)
N Category (No. of Tumor-Involved Regional Lymph Nodes/Presence of In-Transit, Satellite, and/or Microsatellite Metastases)
M Category (Anatomic Site/LDH Level)
Illustration
T = primary tumor; N = regional lymph node; M = distant metastasis; cN = clinical N; CNS = central nervous system; LDH = lactate dehydrogenase; No. = number; p = pathological.
aAdapted from AJCC: Melanoma of the Skin. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 563–85.
bFor example, sentinel lymph node biopsy not performed, regional nodes previously removed for another reason. (Exception: pathological N category is not required for T1 melanomas, use cN).
cPathological stage 0 (melanoma in situ) and T1 do not require pathological evaluation of lymph nodes to complete pathological staging; use cN information to assign their pathological stage.
aClinical trials are an important option for patients with all stages of melanoma because advances in understanding the aberrant molecular and biological pathways have led to rapid drug development. Standard treatment options are available in many clinical trials. Information about ongoing clinical trials is available from the NCI website.
Complete surgical resection of all known disease versus best medical therapy (under clinical evaluation)
Isolated limb perfusion for unresectable extremity melanoma (under clinical evaluation)
Systemic therapy for unresectable disease (under clinical evaluation)
Excision
Surgical excision remains the primary modality for treating localized melanoma. Cutaneous melanomas that have not spread beyond the initial site are highly curable. Localized melanoma is excised with margins proportional to the microstage of the primary lesion.
Standardizing treatment for mucosal melanoma is difficult due to the paucity of prospective data in this rare subgroup. Surgery remains the cornerstone of therapy. Local excision is performed when feasible, as radical resection has not conferred a survival advantage in retrospective studies.[1–3] Optimal excision margins have not been prospectively studied, but the ultimate goal is to obtain negative histological margins.[4]
Lymph node management
Sentinel lymph node biopsy (SLNB)
Lymphatic mapping and SLNB should be considered to assess the presence of occult metastasis in the regional lymph nodes of patients with primary tumors measuring at least 0.8 mm thick with clinically negative nodes. These procedures may identify individuals who can avoid regional lymph node dissection and individuals who may benefit from adjuvant therapy.[5–10]
Multiple studies have demonstrated the diagnostic accuracy of SLNB, with false-negative rates of 0% to 2%.[5,10–15] To ensure accurate identification of the sentinel lymph node, lymphatic mapping and removal of the sentinel lymph node are ideally performed during the same operation as the wide excision of the primary melanoma.
If micrometastatic melanoma is detected, active surveillance with ultrasound of the draining nodal basin is an acceptable treatment recommendation that has widely replaced complete lymph node dissection (CLND). A complete regional lymphadenectomy can be considered in select populations.
For clinically node-negative patients, there is insufficient evidence to define the role of SLNB in sinonasal, anorectal, or vaginal melanoma. However, SLNB has shown feasibility and accuracy in vulvar melanoma.[16,17] Therapeutic dissection is indicated for clinically positive regional nodes in the absence of distant disease.
Complete lymph node dissection (CLND)
Patients can consider CLND for regional control if the sentinel node(s) is microscopically or macroscopically positive.
Adjuvant Therapy
Adjuvant therapy options for patients at high risk of recurrence after complete resection include checkpoint inhibitors and combination signal transduction inhibitor therapy. Ipilimumab was the first checkpoint inhibitor to be approved by the U.S. Food and Drug Administration (FDA) as adjuvant therapy. It has demonstrated improved overall survival (OS) when given at 10 mg/kg (ipi10), compared with placebo (EORTC 18071 [NCT00636168]).[18] However, ipi10 has significant toxicity at this dose. The North American Intergroup Trial E1609 (NCT01274338), designed with three treatment groups, compared ipi10 with ipilimumab at a lower dose of 3 mg/kg (ipi3) (approved for metastatic melanoma) and with high-dose interferon (HDI). Patients who received ipi3 had a significant improvement in OS compared with ipi10.[19] These data do not support HDI as an adjuvant treatment option for melanoma.
Large randomized trials with nivolumab and pembrolizumab and with combination signal transduction inhibitors (dabrafenib plus trametinib) have shown a clinically significant impact on relapse-free survival (RFS). CheckMate 238 (NCT02388906) compared nivolumab with ipi10 and found that nivolumab produced superior RFS and had a more tolerable safety profile.[20] Pembrolizumab produced superior RFS compared with placebo, with data on OS still maturing in the MK-3475-054/KEYNOTE-054 trial (NCT02362594).[21] Dabrafenib plus trametinib produced superior RFS compared with placebo, with data on OS still maturing in the COMBI-AD trial (NCT01682083).[22] Single-agent BRAF-inhibitor therapy with vemurafenib did not show improved RFS compared with placebo in the BRIM8 trial (NCT01667419).[23]
The benefit of immunotherapy with ipilimumab, nivolumab, and pembrolizumab has been seen regardless of programmed death-ligand 1 (PD-L1) expression or the presence of BRAF pathogenic variants. Combination signal transduction inhibitor therapy is an additional option for patients with BRAF variants.
Participation in clinical trials designed to identify treatments that will further extend RFS and OS with less toxicity and shorter treatment schedules is an important option for all patients.
Neoadjuvant Therapy
Neoadjuvant pembrolizumab can be considered for patients with high-risk node-positive disease or resectable metastatic disease based on the results of a phase II trial (SWOG S1801 [NCT03698019]). In the trial, patients who received 1 year of neoadjuvant and adjuvant pembrolizumab had improved event-free survival (EFS) compared with those who underwent primary resection and received adjuvant pembrolizumab.[24] A total of 313 patients with resectable macroscopic stage IIIB to stage IV melanoma were randomly assigned to receive either (1) three cycles of neoadjuvant pembrolizumab (200 mg intravenously [IV] every 3 weeks) followed by surgery and fifteen cycles of adjuvant pembrolizumab or (2) primary surgery followed by eighteen cycles of adjuvant pembrolizumab (200 mg IV every 3 weeks).
At a median follow-up of 15 months, the EFS rate was 72% in patients who received neoadjuvant immunotherapy and 49% in patients who received adjuvant therapy. Neoadjuvant pembrolizumab did not significantly increase toxicity in the perioperative period compared with adjuvant therapy. OS data are not available yet. The FDA has not approved this regimen, pending the completion of a randomized phase III trial.
Systematic Treatment for Unresectable Stage III, Stage IV, and Recurrent Disease
Treatment options for patients with metastatic melanoma have rapidly expanded over the last decade. Two approaches—checkpoint inhibition and targeting the mitogen-activated protein kinase pathway—have improved OS in randomized trials. Given the rapid development of new agents and combinations, patients may consider a clinical trial for initial treatment and at the time of any subsequent progression.
Immunotherapy
Checkpoint inhibitors
Pembrolizumab, nivolumab, ipilimumab, and relatlimab (in a fixed-dose formulation with nivolumab) are checkpoint inhibitors approved by the FDA. Each has demonstrated the ability to impact OS against different comparators in unresectable or advanced disease. Multiple phase III trials are in progress to determine the optimal sequencing of immunotherapies, immunotherapy with targeted therapy, and whether combinations of immunotherapies or immunotherapy plus targeted therapy are superior for increasing OS.
Interleukin-2 (IL-2)
The FDA approved IL-2 in 1998 because of durable complete response rates in a minority of patients (6%–7%) with previously treated metastatic melanoma in eight phase I and II studies. Phase III trials have not been conducted to compare high-dose IL-2 with other treatments or to determine the impact on OS.
Dual checkpoint inhibition
The combination of an anti–programmed death-1 (PD-1) antibody and an anti–cytotoxic T-lymphocyte antigen-4 (CTLA-4) antibody (nivolumab and ipilimumab) has prolonged progression-free survival (PFS) and OS compared with ipilimumab monotherapy. However, this combination is associated with significant toxicity.
Studies have demonstrated a PFS benefit for patients who receive the combination of nivolumab and the anti–lymphocyte-activation gene-3 (LAG-3) antibody relatlimab, compared with nivolumab monotherapy.[25] OS and response rate data are immature.
Signal transduction inhibitors
Studies indicate that both BRAF and MEK inhibitors can significantly impact the natural history of melanoma, although they do not appear to be curative as single agents. Three combination regimens of BRAF and MEK inhibitors have improved PFS and OS compared with BRAF inhibitor monotherapy.
BRAF inhibitors
Vemurafenib
Vemurafenib, approved by the FDA in 2011, has improved PFS and OS in patients with unresectable or advanced disease. Vemurafenib is an orally available, small-molecule, selective BRAF V600E kinase inhibitor, and its indication is limited to patients with a demonstrated BRAF V600E pathogenic variant by an FDA-approved test.[15]
Dabrafenib
Dabrafenib is an orally available, small-molecule, selective BRAF inhibitor that was approved by the FDA in 2013. An international multicenter trial (BREAK-3 [NCT01227889]) showed that dabrafenib improved PFS when compared with dacarbazine.[26]
Encorafenib
Encorafenib is an orally available, small-molecule, selective BRAF inhibitor. The FDA approved encorafenib in 2018 in combination with the MEK inhibitor binimetinib. A phase III randomized study demonstrated that encorafenib improved PFS and OS when compared with vemurafenib monotherapy.[27]
MEK inhibitors
Trametinib
Trametinib is an orally available, small-molecule, selective inhibitor of MEK1 and MEK2. The FDA approved trametinib in 2013 for patients with unresectable or metastatic melanoma with BRAF V600E or V600K pathogenic variants. Trametinib demonstrated improved PFS when compared with dacarbazine.[28]
Cobimetinib
Cobimetinib is an orally available, small-molecule, selective MEK inhibitor. The FDA approved cobimetinib in 2015 for use in combination with the BRAF inhibitor vemurafenib. For more information, visit the Combination signal transduction inhibitor therapy section.
Binimetinib
Binimetinib is an orally available, small-molecule, selective MEK1 and MEK2 inhibitor. The FDA approved binimetinib in 2018 for use in combination with the BRAF inhibitor encorafenib.
c-KIT inhibitors
Early data suggest that mucosal or acral melanomas with activating pathogenic variants or amplifications in KIT may be sensitive to a variety of c-KIT inhibitors.[29–31] Phase II and phase III trials are available for patients with unresectable stage III or stage IV melanoma and a KIT pathogenic variant.
Combination signal transduction inhibitor therapy
The FDA approved the combination regimens dabrafenib plus trametinib, vemurafenib plus cobimetinib, and encorafenib plus binimetinib in patients with unresectable or metastatic melanomas that carry BRAF V600E or V600K pathogenic variants as confirmed by an FDA-approved test. The approvals were based on improved PFS and OS when compared with a single-agent BRAF inhibitor (either dabrafenib or vemurafenib).
Combination signal transduction inhibitor therapy plus anti–PD-L1 therapy
The triplet regimen of cobimetinib (MEK inhibitor), vemurafenib (BRAF kinase inhibitor), and atezolizumab (PD-L1 inhibitor) is an FDA-approved regimen. A phase III study showed improved PFS over the combination of cobimetinib and vemurafenib.[32] However, there was not a significant difference in OS between the arms of the study and there was a higher rate of toxicities in the combination arm. As such, this regimen is not commonly used.
Chemotherapy
Dacarbazine
Dacarbazine was approved in 1970 based on overall response rates. Phase III trials indicated an overall response rate of 10% to 20%, with rare complete responses observed. An impact on OS has not been demonstrated in randomized trials.[33–36] When used as a control arm for registration trials of ipilimumab and vemurafenib in previously untreated patients with metastatic melanoma, dacarbazine was shown to be inferior for OS.
Temozolomide
Temozolomide, an oral alkylating agent, appeared to be similar to IV dacarbazine in a randomized phase III trial with a primary end point of OS. However, because the trial was designed to demonstrate the superiority of temozolomide, which was not achieved, the trial was left with a sample size that was inadequate to provide statistical proof of noninferiority.[34]
Palliative local therapy
Regional lymphadenectomy may be used as palliative care for melanoma that is metastatic to distant, lymph node–bearing areas. Resection may be used as palliative care for isolated metastases to the lung, gastrointestinal tract, bone, or sometimes the brain, with occasional long-term survival.[20,37,38]
References
Moreno MA, Roberts DB, Kupferman ME, et al.: Mucosal melanoma of the nose and paranasal sinuses, a contemporary experience from the M. D. Anderson Cancer Center. Cancer 116 (9): 2215-23, 2010. [PUBMED Abstract]
Iddings DM, Fleisig AJ, Chen SL, et al.: Practice patterns and outcomes for anorectal melanoma in the USA, reviewing three decades of treatment: is more extensive surgical resection beneficial in all patients? Ann Surg Oncol 17 (1): 40-4, 2010. [PUBMED Abstract]
Sugiyama VE, Chan JK, Shin JY, et al.: Vulvar melanoma: a multivariable analysis of 644 patients. Obstet Gynecol 110 (2 Pt 1): 296-301, 2007. [PUBMED Abstract]
Shen P, Wanek LA, Morton DL: Is adjuvant radiotherapy necessary after positive lymph node dissection in head and neck melanomas? Ann Surg Oncol 7 (8): 554-9; discussion 560-1, 2000. [PUBMED Abstract]
Hochwald SN, Coit DG: Role of elective lymph node dissection in melanoma. Semin Surg Oncol 14 (4): 276-82, 1998. [PUBMED Abstract]
Wagner JD, Gordon MS, Chuang TY, et al.: Current therapy of cutaneous melanoma. Plast Reconstr Surg 105 (5): 1774-99; quiz 1800-1, 2000. [PUBMED Abstract]
Cascinelli N, Morabito A, Santinami M, et al.: Immediate or delayed dissection of regional nodes in patients with melanoma of the trunk: a randomised trial. WHO Melanoma Programme. Lancet 351 (9105): 793-6, 1998. [PUBMED Abstract]
Koops HS, Vaglini M, Suciu S, et al.: Prophylactic isolated limb perfusion for localized, high-risk limb melanoma: results of a multicenter randomized phase III trial. European Organization for Research and Treatment of Cancer Malignant Melanoma Cooperative Group Protocol 18832, the World Health Organization Melanoma Program Trial 15, and the North American Perfusion Group Southwest Oncology Group-8593. J Clin Oncol 16 (9): 2906-12, 1998. [PUBMED Abstract]
Wong SL, Balch CM, Hurley P, et al.: Sentinel lymph node biopsy for melanoma: American Society of Clinical Oncology and Society of Surgical Oncology joint clinical practice guideline. J Clin Oncol 30 (23): 2912-8, 2012. [PUBMED Abstract]
Kirkwood JM, Strawderman MH, Ernstoff MS, et al.: Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14 (1): 7-17, 1996. [PUBMED Abstract]
Kirkwood JM, Ibrahim JG, Sondak VK, et al.: High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol 18 (12): 2444-58, 2000. [PUBMED Abstract]
Eggermont AM, Suciu S, Santinami M, et al.: Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet 372 (9633): 117-26, 2008. [PUBMED Abstract]
Hancock BW, Wheatley K, Harris S, et al.: Adjuvant interferon in high-risk melanoma: the AIM HIGH Study–United Kingdom Coordinating Committee on Cancer Research randomized study of adjuvant low-dose extended-duration interferon Alfa-2a in high-risk resected malignant melanoma. J Clin Oncol 22 (1): 53-61, 2004. [PUBMED Abstract]
Chapman PB, Hauschild A, Robert C, et al.: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364 (26): 2507-16, 2011. [PUBMED Abstract]
Leitao MM, Cheng X, Hamilton AL, et al.: Gynecologic Cancer InterGroup (GCIG) consensus review for vulvovaginal melanomas. Int J Gynecol Cancer 24 (9 Suppl 3): S117-22, 2014. [PUBMED Abstract]
Trifirò G, Travaini LL, Sanvito F, et al.: Sentinel node detection by lymphoscintigraphy and sentinel lymph node biopsy in vulvar melanoma. Eur J Nucl Med Mol Imaging 37 (4): 736-41, 2010. [PUBMED Abstract]
Eggermont AM, Chiarion-Sileni V, Grob JJ, et al.: Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N Engl J Med 375 (19): 1845-1855, 2016. [PUBMED Abstract]
Tarhini AA, Lee SJ, Hodi FS, et al.: Phase III Study of Adjuvant Ipilimumab (3 or 10 mg/kg) Versus High-Dose Interferon Alfa-2b for Resected High-Risk Melanoma: North American Intergroup E1609. J Clin Oncol 38 (6): 567-575, 2020. [PUBMED Abstract]
Leo F, Cagini L, Rocmans P, et al.: Lung metastases from melanoma: when is surgical treatment warranted? Br J Cancer 83 (5): 569-72, 2000. [PUBMED Abstract]
Eggermont AMM, Blank CU, Mandala M, et al.: Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N Engl J Med 378 (19): 1789-1801, 2018. [PUBMED Abstract]
Long GV, Hauschild A, Santinami M, et al.: Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N Engl J Med 377 (19): 1813-1823, 2017. [PUBMED Abstract]
Maio M, Lewis K, Demidov L, et al.: Adjuvant vemurafenib in resected, BRAFV600 mutation-positive melanoma (BRIM8): a randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol 19 (4): 510-520, 2018. [PUBMED Abstract]
Patel SP, Othus M, Chen Y, et al.: Neoadjuvant-Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N Engl J Med 388 (9): 813-823, 2023. [PUBMED Abstract]
Tawbi HA, Schadendorf D, Lipson EJ, et al.: Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N Engl J Med 386 (1): 24-34, 2022. [PUBMED Abstract]
Hauschild A, Grob JJ, Demidov LV, et al.: Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380 (9839): 358-65, 2012. [PUBMED Abstract]
Dummer R, Ascierto PA, Gogas HJ, et al.: Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 19 (10): 1315-1327, 2018. [PUBMED Abstract]
Flaherty KT, Robert C, Hersey P, et al.: Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367 (2): 107-14, 2012. [PUBMED Abstract]
Hodi FS, Friedlander P, Corless CL, et al.: Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol 26 (12): 2046-51, 2008. [PUBMED Abstract]
Guo J, Si L, Kong Y, et al.: Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 29 (21): 2904-9, 2011. [PUBMED Abstract]
Carvajal RD, Antonescu CR, Wolchok JD, et al.: KIT as a therapeutic target in metastatic melanoma. JAMA 305 (22): 2327-34, 2011. [PUBMED Abstract]
Gutzmer R, Stroyakovskiy D, Gogas H, et al.: Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 395 (10240): 1835-1844, 2020. [PUBMED Abstract]
Chapman PB, Einhorn LH, Meyers ML, et al.: Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol 17 (9): 2745-51, 1999. [PUBMED Abstract]
Middleton MR, Grob JJ, Aaronson N, et al.: Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 18 (1): 158-66, 2000. [PUBMED Abstract]
Avril MF, Aamdal S, Grob JJ, et al.: Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: a phase III study. J Clin Oncol 22 (6): 1118-25, 2004. [PUBMED Abstract]
Robert C, Thomas L, Bondarenko I, et al.: Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364 (26): 2517-26, 2011. [PUBMED Abstract]
Ollila DW, Hsueh EC, Stern SL, et al.: Metastasectomy for recurrent stage IV melanoma. J Surg Oncol 71 (4): 209-13, 1999. [PUBMED Abstract]
Gutman H, Hess KR, Kokotsakis JA, et al.: Surgery for abdominal metastases of cutaneous melanoma. World J Surg 25 (6): 750-8, 2001. [PUBMED Abstract]
There is no high-level evidence to guide the recommended excision margins for stage 0 (or in situ) melanoma. Consensus guidelines recommend margins of at least 5 mm for stage 0 melanoma, with a goal of achieving microscopically negative margins. However, 5 mm margins may be inadequate for some cases of in situ melanoma, and wider margins may be required.[1,2]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Kunishige JH, Doan L, Brodland DG, et al.: Comparison of surgical margins for lentigo maligna versus melanoma in situ. J Am Acad Dermatol 81 (1): 204-212, 2019. [PUBMED Abstract]
Ellison PM, Zitelli JA, Brodland DG: Mohs micrographic surgery for melanoma: A prospective multicenter study. J Am Acad Dermatol 81 (3): 767-774, 2019. [PUBMED Abstract]
Treatment of Stage IA Melanoma
Treatment Options for Stage IA Melanoma
Treatment options for stage IA (pT1a or pT1b) melanoma include:
No randomized controlled trials have assessed only melanomas that are less than 1 mm thick. Evidence from randomized controlled clinical trials that included patients with melanomas of this size suggests that they may be adequately treated with radial excision margins of 1 cm.
Evidence (excision):
A randomized trial that compared narrow margins (1 cm) with wide margins (≥3 cm) in 612 patients with melanomas no thicker than 2 mm included 359 patients with melanomas measuring 1 mm or less.[1,2][Level of evidence A1]
No difference was observed between the two groups in the development of metastatic disease, disease-free survival (DFS), or overall survival (OS).
There were no local recurrences in patients with melanomas measuring 1 mm or less in either cohort.
A Swedish multicenter study included 989 patients with primary melanomas located on the trunk or extremities with a tumor thickness between 0.8 mm and 2 mm. Patients were randomly assigned to undergo wide excision with margins of 2 cm (n = 476) or 5 cm (n = 513).[3][Level of evidence A1]
With a median follow-up of 11 years, there was no statistically significant difference in OS between the two groups.
With a median follow-up of 8 years, there was no statistically significant difference in recurrence-free survival between the two groups.
The local recurrence rate was 1%, and there was no significant difference between the treatment arms.
A European, multicenter, randomized trial compared margins of 2 cm (n = 161) versus 5 cm (n = 165) in 326 patients with primary melanomas with a thickness of 2.1 mm or less. The study included 141 patients with melanomas measuring 1 mm or less. [3,4]Level of evidence A1]
There was no statistically significant difference in 10-year DFS or OS between the two groups.
Local recurrence occurred in one patient treated with a 2-cm margin and four patients treated with 5-cm margins.
Sentinel lymph node biopsy
Lymphatic mapping and SLNB for patients with high-risk, thin melanomas (≥0.8 mm) or ulcerated lesions measuring less than 0.8 mm may identify individuals with occult nodal disease. These procedures should be considered, particularly if other adverse prognostic features are present. Patients with clinically occult regional nodal metastases may benefit from ultrasound surveillance of regional lymph nodes and adjuvant therapy.[5–10]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Veronesi U, Cascinelli N: Narrow excision (1-cm margin). A safe procedure for thin cutaneous melanoma. Arch Surg 126 (4): 438-41, 1991. [PUBMED Abstract]
Veronesi U, Cascinelli N, Adamus J, et al.: Thin stage I primary cutaneous malignant melanoma. Comparison of excision with margins of 1 or 3 cm. N Engl J Med 318 (18): 1159-62, 1988. [PUBMED Abstract]
Cohn-Cedermark G, Rutqvist LE, Andersson R, et al.: Long term results of a randomized study by the Swedish Melanoma Study Group on 2-cm versus 5-cm resection margins for patients with cutaneous melanoma with a tumor thickness of 0.8-2.0 mm. Cancer 89 (7): 1495-501, 2000. [PUBMED Abstract]
Balch CM, Soong SJ, Smith T, et al.: Long-term results of a prospective surgical trial comparing 2 cm vs. 4 cm excision margins for 740 patients with 1-4 mm melanomas. Ann Surg Oncol 8 (2): 101-8, 2001. [PUBMED Abstract]
Essner R, Conforti A, Kelley MC, et al.: Efficacy of lymphatic mapping, sentinel lymphadenectomy, and selective complete lymph node dissection as a therapeutic procedure for early-stage melanoma. Ann Surg Oncol 6 (5): 442-9, 1999 Jul-Aug. [PUBMED Abstract]
Gershenwald JE, Thompson W, Mansfield PF, et al.: Multi-institutional melanoma lymphatic mapping experience: the prognostic value of sentinel lymph node status in 612 stage I or II melanoma patients. J Clin Oncol 17 (3): 976-83, 1999. [PUBMED Abstract]
Mraz-Gernhard S, Sagebiel RW, Kashani-Sabet M, et al.: Prediction of sentinel lymph node micrometastasis by histological features in primary cutaneous malignant melanoma. Arch Dermatol 134 (8): 983-7, 1998. [PUBMED Abstract]
Morton DL, Thompson JF, Cochran AJ, et al.: Sentinel-node biopsy or nodal observation in melanoma. N Engl J Med 355 (13): 1307-17, 2006. [PUBMED Abstract]
Faries MB, Thompson JF, Cochran AJ, et al.: Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N Engl J Med 376 (23): 2211-2222, 2017. [PUBMED Abstract]
Leiter U, Stadler R, Mauch C, et al.: Complete lymph node dissection versus no dissection in patients with sentinel lymph node biopsy positive melanoma (DeCOG-SLT): a multicentre, randomised, phase 3 trial. Lancet Oncol 17 (6): 757-767, 2016. [PUBMED Abstract]
Treatment of Stage IB Melanoma
Treatment Options for Stage IB Melanoma
Treatment options for stage IB (pT2a) melanoma include:
No randomized controlled trials have compared 1-cm margins with 2-cm margins for melanomas measuring 2 mm or thinner. Evidence suggests that lesions no thicker than 2 mm may be treated conservatively with clinical radial excision margins of 1 cm to 2 cm. The decision to pursue a 1-cm versus a 2-cm margin should be based on patient and lesion factors, including functional and cosmetic limitations.
Evidence (excision):
A randomized trial compared narrow margins (1 cm) with wide margins (≥3 cm) in 612 patients with melanomas no thicker than 2 mm.[1,2][Level of evidence A1]
No difference was observed between the two groups in the development of metastatic disease, disease-free survival (DFS), or overall survival (OS).
The risk of recurrence was 2.7% in patients with melanomas thicker than 1 mm who underwent narrow margins excision. No local recurrences occurred in the wider margin cohort.
A Swedish multicenter study included 989 patients with primary melanomas located on the trunk or extremities with a tumor thickness between 0.8 mm and 2 mm. Patients were randomly assigned to undergo wide excision with margins of either 2 cm (n = 476) or 5 cm (n = 513).[3][Level of evidence A1]
With a median follow-up of 11 years, there was no statistically significant difference in OS between the two treatment groups.
With a median follow-up of 8 years, there was no statistically significant difference in recurrence-free survival between the two groups.
The local recurrence rate was 1%, and there was no significant difference between the treatment arms.
A European, multicenter, randomized trial compared margins of 2 cm (n = 161) versus 5 cm (n = 165) in 326 patients with primary melanomas with a thickness of 2.1 mm or less. The study included 141 patients with melanomas measuring 1 mm or less.[4][Level of evidence A1]
There was no statistically significant difference in 10-year DFS or OS between the two treatment groups.
Local recurrence occurred in one patient treated with a 2-cm margin and four patients treated with 5-cm margins.
The Intergroup Melanoma Surgical Trial compared radial excision margins of 2 cm versus 4 cm in patients with melanomas of 1 mm to 4 mm in thickness.[5][Level of evidence A1]
With a median follow-up of 10 years, there was no significant difference in OS, disease-specific survival, or local recurrence between the two treatment groups.
A single-center retrospective study included 2,131 patients with melanomas between 1 mm and 2 mm (pT2) in thickness. The study compared excision with histological margins of 8 mm versus 16 mm (corresponding to 1-cm versus 2-cm clinical margins).[6][Level of evidence C1]
There was no significant difference in 5-year melanoma-specific survival (P = .210) or 5-year DFS (P = .202) between the two groups.
On multivariate analysis, peripheral excision margins did not influence local or in-transit recurrence.
Another single-center retrospective study included 576 patients with melanomas of 1 mm to 2 mm (pT2) in thickness. The study compared excision with clinical margins of 1 cm versus 2 cm .[7][Level of evidence C2]
The local recurrence rate was significantly higher in the 1-cm margin group than in the 2-cm margin group (3.6% vs. 0.9%; P = .044) on univariate analysis, but the rate was no longer significantly different on multivariate analysis.
There was no difference in OS between the two groups.
Lymph node management
Elective regional lymph node dissection has no proven benefit for patients with stage I melanoma.[8]
Lymphatic mapping and sentinel lymph node biopsy (SLNB) for patients who have tumors of intermediate thickness may identify individuals with occult nodal disease. Patients with clinically occult regional nodal metastases may benefit from ultrasound surveillance of regional lymph nodes and adjuvant therapy.[9–14]
Evidence (SLNB versus observation):
The International Multicenter Selective Lymphadenectomy Trial (MSLT-1) included 1,269 patients with intermediate-thickness (defined as 1.2–3.5 mm in this study) primary melanomas. The primary end point was melanoma-specific survival.[15][Level of evidence A1]
At a median follow-up of 59.8 months, there was no melanoma-specific survival advantage for patients randomly assigned to undergo wide excision plus SLNB, followed by immediate completion lymphadenectomy for node positivity versus nodal observation and delayed lymphadenectomy for subsequent nodal recurrence.
This trial was not designed to detect a difference in the impact of lymphadenectomy in patients with microscopic lymph node involvement.
Evidence (completion lymphadenectomy vs. observation with serial ultrasound of draining nodal basin):
The Multicenter Selective Lymphadenectomy Trial II (MSLT-II [NCT00297895]) included 1,934 patients with primary melanomas and a positive sentinel node(s). Patients were randomly assigned to undergo either completion lymphadenectomy (n = 967) or nodal observation with ultrasound (n = 967). The primary end point was melanoma-specific survival.[13][Level of evidence A1]
At a follow-up of 3 years, there was no melanoma-specific survival advantage for patients randomly assigned to undergo either completion lymphadenectomy versus nodal observation with ultrasound and delayed lymphadenectomy for subsequent nodal recurrence (86% [±1.3%] vs. 86% [±1.2%]; hazard ratio [HR], 1.08; 95% confidence interval [CI], 0.88–1.34; P = .42).
At 3 years, the rate of nodal recurrence was 69% lower in the dissection group than in the observation group, with regional nodal disease control rates of 92% (±1.0%) versus 77% (±1.5%), respectively (HR, 0.31; 95% CI, 0.24–0.41; P < .001).
With a median follow-up of 43 months, there was no significant difference in distant metastasis–free survival (DMFS) between the two groups (HR, 1.10; 95% CI, 0.92–1.31; P = .31).
Adverse events were more common among the surgical cohort. Specifically, lymphedema occurred in 24.1% of patients in the dissection group versus 6.3% of patients in the observation group (P < .0001).
A subgroup analysis did not identify any group (i.e., patients with >1 positive sentinel lymph node or a sentinel lymph node tumor >1 mm in diameter) that would benefit from a complete lymph node dissection.
The DeCOG-SLT trial (NCT02434107) included 483 patients with primary melanoma and microscopically detected nodal metastases. Patients were randomly assigned to undergo either completion lymphadenectomy (n = 240) or observation (n = 233). The primary end point was DMFS.[14][Level of evidence B1]
At a median follow-up of 35.5 months, there was no difference in DMFS in patients randomly assigned to undergo observation (77%; 90% CI, 71.9%–82.1%) versus completion lymphadenectomy (74.9%; 90% CI, 69.5%–80.3%) (HR, 1.03; 90% CI 0.71-1.50, P = 0.87).
At 3 years, the OS rates were similar between patients randomly assigned to undergo observation (81.7%; 90% CI, 76.8%–86.6%) versus completion lymphadenectomy (81.2%; 90% CI, 76.1%–86.3%) (HR, 0.96; 90% CI, 0.67–1.38; P = .87), although the trial was closed early due to low event rates.
In an exploratory analysis of DMFS in patients with micrometastasis thicker than 1 mm, there was no difference between the treatment groups (HR, 1.03; 90% CI, 0.63–1.71).
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Veronesi U, Cascinelli N: Narrow excision (1-cm margin). A safe procedure for thin cutaneous melanoma. Arch Surg 126 (4): 438-41, 1991. [PUBMED Abstract]
Veronesi U, Cascinelli N, Adamus J, et al.: Thin stage I primary cutaneous malignant melanoma. Comparison of excision with margins of 1 or 3 cm. N Engl J Med 318 (18): 1159-62, 1988. [PUBMED Abstract]
Cohn-Cedermark G, Rutqvist LE, Andersson R, et al.: Long term results of a randomized study by the Swedish Melanoma Study Group on 2-cm versus 5-cm resection margins for patients with cutaneous melanoma with a tumor thickness of 0.8-2.0 mm. Cancer 89 (7): 1495-501, 2000. [PUBMED Abstract]
Khayat D, Rixe O, Martin G, et al.: Surgical margins in cutaneous melanoma (2 cm versus 5 cm for lesions measuring less than 2.1-mm thick). Cancer 97 (8): 1941-6, 2003. [PUBMED Abstract]
Balch CM, Urist MM, Karakousis CP, et al.: Efficacy of 2-cm surgical margins for intermediate-thickness melanomas (1 to 4 mm). Results of a multi-institutional randomized surgical trial. Ann Surg 218 (3): 262-7; discussion 267-9, 1993. [PUBMED Abstract]
Haydu LE, Stollman JT, Scolyer RA, et al.: Minimum Safe Pathologic Excision Margins for Primary Cutaneous Melanomas (1-2 mm in Thickness): Analysis of 2131 Patients Treated at a Single Center. Ann Surg Oncol 23 (4): 1071-81, 2016. [PUBMED Abstract]
Hudson LE, Maithel SK, Carlson GW, et al.: 1 or 2 cm margins of excision for T2 melanomas: do they impact recurrence or survival? Ann Surg Oncol 20 (1): 346-51, 2013. [PUBMED Abstract]
Hochwald SN, Coit DG: Role of elective lymph node dissection in melanoma. Semin Surg Oncol 14 (4): 276-82, 1998. [PUBMED Abstract]
Essner R, Conforti A, Kelley MC, et al.: Efficacy of lymphatic mapping, sentinel lymphadenectomy, and selective complete lymph node dissection as a therapeutic procedure for early-stage melanoma. Ann Surg Oncol 6 (5): 442-9, 1999 Jul-Aug. [PUBMED Abstract]
Gershenwald JE, Thompson W, Mansfield PF, et al.: Multi-institutional melanoma lymphatic mapping experience: the prognostic value of sentinel lymph node status in 612 stage I or II melanoma patients. J Clin Oncol 17 (3): 976-83, 1999. [PUBMED Abstract]
Mraz-Gernhard S, Sagebiel RW, Kashani-Sabet M, et al.: Prediction of sentinel lymph node micrometastasis by histological features in primary cutaneous malignant melanoma. Arch Dermatol 134 (8): 983-7, 1998. [PUBMED Abstract]
Morton DL, Thompson JF, Cochran AJ, et al.: Sentinel-node biopsy or nodal observation in melanoma. N Engl J Med 355 (13): 1307-17, 2006. [PUBMED Abstract]
Faries MB, Thompson JF, Cochran AJ, et al.: Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N Engl J Med 376 (23): 2211-2222, 2017. [PUBMED Abstract]
Leiter U, Stadler R, Mauch C, et al.: Complete lymph node dissection versus no dissection in patients with sentinel lymph node biopsy positive melanoma (DeCOG-SLT): a multicentre, randomised, phase 3 trial. Lancet Oncol 17 (6): 757-767, 2016. [PUBMED Abstract]
Morton DL, Thompson JF, Cochran AJ, et al.: Final trial report of sentinel-node biopsy versus nodal observation in melanoma. N Engl J Med 370 (7): 599-609, 2014. [PUBMED Abstract]
Evidence suggests that lesions no thicker than 2 mm (pT2b) may be treated conservatively with clinical radial excision margins of 1 cm to 2 cm. The decision to pursue a 1-cm versus 2-cm margin should be based on patient and lesion factors, including functional and cosmetic limitations. Evidence suggests that for melanomas measuring 2 mm or thicker (pT3a/b, pT4a/b), clinical radial margins of 2 cm are recommended. There is no evidence to support that a wider margin is beneficial.[1]
Evidence (excision):
The Intergroup Melanoma Surgical Trial Task 2b compared 2-cm versus 4-cm margins for 740 patients with melanomas that were 1 mm to 4 mm thick.[2]
With a median follow-up of more than 10 years, no significant difference in local recurrence or survival was observed between the two groups.
The reduction in margins from 4 cm to 2 cm was associated with a statistically significant reduction in the need for skin grafting (from 46% to 11%; P < .001).
A study conducted in the United Kingdom randomly assigned patients with melanomas thicker than 2 mm to undergo excision with either 1-cm or 3-cm margins.[3,4][Level of evidence A1]
At a median follow-up of 5 years, patients who underwent excision with 1-cm margins had higher rates of locoregional recurrence (hazard ratio [HR], 1.34; 95% confidence interval [CI], 1.06–1.71; P = .02).
No difference in overall survival (OS) was seen (HR, 0.81; 95% CI, 0.58–1.13; P = .386) between the two groups. However, at 8.8 years, there was a melanoma-specific survival advantage for patients who underwent excision with 3-cm margins compared with 1-cm margins (HR, 1.24; 95% CI, 1.01–1.52; P = .036).
This study suggests that 1-cm margins may not be adequate for patients with melanomas thicker than 2 mm.
In a multicenter international trial (NCT03638492), the Swedish and Danish melanoma groups randomly assigned patients with localized cutaneous melanoma thicker than 2mm to undergo excision with clinical margins of either 2 cm (n = 471) or 4 cm (n = 465).[1][Level of evidence A1]
With a median follow-up of 19.6 years, no significant difference in OS or melanoma-specific survival was observed between the two groups (HR, 0.98; 95% CI, 0.83–1.14; P = .75).
Lymph node management
Lymphatic mapping and sentinel lymph node biopsy (SLNB)
Lymphatic mapping and SLNB have assessed the presence of occult metastasis in the regional lymph nodes of patients with stage II disease. These procedures may identify individuals who can avoid regional lymph node dissection and individuals who may benefit from adjuvant therapy.[5–9]
To ensure accurate identification of the sentinel lymph node, lymphatic mapping and removal of the sentinel lymph node are performed during the same operation as the wide excision of the primary melanoma.
With the use of a vital blue dye and a radiopharmaceutical agent injected at the site of the primary tumor, the first lymph node in the lymphatic basin that drains the lesion can be identified, removed, and examined microscopically. Multiple studies have demonstrated the diagnostic accuracy of SLNB, with false-negative rates of 0% to 2%.[5,10–14]
Regional lymphadenectomy
In patients with microscopic melanoma in regional lymph nodes, immediate completion lymphadenectomy has widely been replaced by active observation, as long as close follow-up with nodal ultrasound surveillance can be achieved.[15,16] Completion lymphadenectomy may still be considered on a case-by-case basis.
Evidence (completion lymphadenectomy vs. observation with serial ultrasound of draining nodal basin):
The Multicenter Selective Lymphadenectomy Trial II (MSLT-II [NCT00297895]) included 1,934 patients with primary melanomas and a positive sentinel node(s). Patients were randomly assigned to undergo either completion lymphadenectomy (n = 967) or nodal observation with ultrasound (n = 967). The primary end point was melanoma-specific survival.[15][Level of evidence A1]
After a follow-up of 3 years, there was no melanoma-specific survival advantage for patients randomly assigned to undergo completion lymphadenectomy versus nodal observation with ultrasound and delayed lymphadenectomy for subsequent nodal recurrence (86% [±1.3%] vs. 86% [±1.2%]; HR, 1.08; 95% CI, 0.88–1.34; P = .42).
At 3 years, the rate of nodal recurrence was 69% lower in the dissection group than in the observation group, with regional nodal disease control rates of 92% (±1.0%) versus 77% (±1.5%), respectively (HR, 0.31; 95% CI, 0.24–0.41; P < .001).
With a median follow-up of 43 months, there was no significant difference in distant metastasis–free survival (DMFS) between the two groups (HR, 1.10; 95% CI, 0.92–1.31; P = .31).
Adverse events were more common among the surgical cohort. Specifically, lymphedema occurred in 24.1% of patients in the dissection group versus 6.3% of patients in the observation group (P < .0001).
A subgroup analysis did not identify any group (i.e., patients with >1 positive sentinel lymph node or a sentinel lymph node tumor diameter >1 mm) that would benefit from a complete lymph node dissection.
The DeCOG-SLT trial (NCT02434107) included 483 patients with primary melanoma and microscopically detected nodal metastases. Patients were randomly assigned to undergo completion lymphadenectomy (n = 240) or observation (n = 233). The primary end point was DMFS.[16][Level of evidence B1]
At a median follow-up of 35.5 months, there was no difference in DMFS between patients randomly assigned to undergo observation (77%; 90% CI, 71.9%–82.1%) versus completion lymphadenectomy (74.9%; 90% CI, 69.5%–80.3%) (HR, 1.03; 90% CI, 0.71–1.50; P = .87).
At 3 years, the OS rates were similar between patients randomly assigned to undergo observation (81.7%; 90% CI, 76.8%–86.6%) versus completion lymphadenectomy (81.2%; 90% CI, 76.1%–86.3%) (HR, 0.96; 90% CI, 0.67–1.38; P = .87), although the trial was closed early due to low event rates.
In an exploratory analysis of DMFS in patients with micrometastasis thicker than 1 mm, there was no difference between the treatment groups (HR, 1.03; 90% CI, 0.63–1.71).
Adjuvant therapy
Adjuvant therapeutic options are expanding for patients at high risk of recurrence after complete resection. Patients with resected stage IIB or stage IIC melanoma, despite not having lymph node involvement, have a similar risk of recurrence and melanoma-specific death as patients with stage III melanoma. As outlined in the Treatment of Resectable Stage III Melanoma section, the U.S. Food and Drug Administration (FDA) has approved several agents as adjuvant therapy for patients with resected stage III melanoma.
Immunotherapy
Adjuvant immunotherapy has demonstrated a recurrence-free survival (RFS) and DMFS benefit in patients with high-risk stage II melanoma that has been resected. These results led to FDA approval for the anti–programmed death-1 agent pembrolizumab. Data regarding potential OS benefit are still pending.
Checkpoint inhibitors
Pembrolizumab
Evidence (pembrolizumab):
In a multinational double-blind trial (KEYNOTE-716 [NCT03553836]), patients with completely resected stage IIB or stage IIC melanoma were randomly assigned (1:1) to receive either pembrolizumab or placebo. The primary end point was RFS, defined as the time from randomization until the date of first recurrence or death from any cause. If recurrence was documented, patients could cross over or repeat treatment with pembrolizumab. The secondary end point was DMFS. Pembrolizumab was given as an intravenous infusion of 200 mg every 3 weeks, for a total of 17 doses (approximately 1 year).
A total of 976 patients were randomly assigned: 487 to pembrolizumab and 489 to placebo. Baseline characteristics were balanced, including the number of patients with ulcerated tumors (40% of stage IIB; 36% of stage IIC) in both arms.[17]
At the first interim analysis with a median follow-up of 14.4 months, the 12-month RFS rate was 90.0% (95% CI, 87%–93%) in the pembrolizumab group and 83% (95% CI, 79%–86%) in the placebo group.[17][Level of evidence B1]
At the second interim analysis with a median follow-up of 20.9 months, the 18-month RFS rate was 86% (95% CI, 82%–89%) in the pembrolizumab group and 77% (95% CI, 73%–81%) in the placebo group.
At a median follow-up of 39.4 months, the 36-month RFS rate was 76.2% in the pembrolizumab group and 63.4% in the placebo group. An updated analysis of DMFS reported a 36-month DMFS rate of 84.4% in the pembrolizumab group and 74.7% in the placebo group. Median DMFS was not reached in either group.[18,19]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Utjés D, Malmstedt J, Teras J, et al.: 2-cm versus 4-cm surgical excision margins for primary cutaneous melanoma thicker than 2 mm: long-term follow-up of a multicentre, randomised trial. Lancet 394 (10197): 471-477, 2019. [PUBMED Abstract]
Balch CM, Urist MM, Karakousis CP, et al.: Efficacy of 2-cm surgical margins for intermediate-thickness melanomas (1 to 4 mm). Results of a multi-institutional randomized surgical trial. Ann Surg 218 (3): 262-7; discussion 267-9, 1993. [PUBMED Abstract]
Thomas JM, Newton-Bishop J, A’Hern R, et al.: Excision margins in high-risk malignant melanoma. N Engl J Med 350 (8): 757-66, 2004. [PUBMED Abstract]
Hayes AJ, Maynard L, Coombes G, et al.: Wide versus narrow excision margins for high-risk, primary cutaneous melanomas: long-term follow-up of survival in a randomised trial. Lancet Oncol 17 (2): 184-192, 2016. [PUBMED Abstract]
Gershenwald JE, Thompson W, Mansfield PF, et al.: Multi-institutional melanoma lymphatic mapping experience: the prognostic value of sentinel lymph node status in 612 stage I or II melanoma patients. J Clin Oncol 17 (3): 976-83, 1999. [PUBMED Abstract]
McMasters KM, Reintgen DS, Ross MI, et al.: Sentinel lymph node biopsy for melanoma: controversy despite widespread agreement. J Clin Oncol 19 (11): 2851-5, 2001. [PUBMED Abstract]
Cherpelis BS, Haddad F, Messina J, et al.: Sentinel lymph node micrometastasis and other histologic factors that predict outcome in patients with thicker melanomas. J Am Acad Dermatol 44 (5): 762-6, 2001. [PUBMED Abstract]
Essner R: The role of lymphoscintigraphy and sentinel node mapping in assessing patient risk in melanoma. Semin Oncol 24 (1 Suppl 4): S8-10, 1997. [PUBMED Abstract]
Chan AD, Morton DL: Sentinel node detection in malignant melanoma. Recent Results Cancer Res 157: 161-77, 2000. [PUBMED Abstract]
Morton DL, Wen DR, Wong JH, et al.: Technical details of intraoperative lymphatic mapping for early stage melanoma. Arch Surg 127 (4): 392-9, 1992. [PUBMED Abstract]
Reintgen D, Cruse CW, Wells K, et al.: The orderly progression of melanoma nodal metastases. Ann Surg 220 (6): 759-67, 1994. [PUBMED Abstract]
Thompson JF, McCarthy WH, Bosch CM, et al.: Sentinel lymph node status as an indicator of the presence of metastatic melanoma in regional lymph nodes. Melanoma Res 5 (4): 255-60, 1995. [PUBMED Abstract]
Uren RF, Howman-Giles R, Thompson JF, et al.: Lymphoscintigraphy to identify sentinel lymph nodes in patients with melanoma. Melanoma Res 4 (6): 395-9, 1994. [PUBMED Abstract]
Bostick P, Essner R, Glass E, et al.: Comparison of blue dye and probe-assisted intraoperative lymphatic mapping in melanoma to identify sentinel nodes in 100 lymphatic basins. Arch Surg 134 (1): 43-9, 1999. [PUBMED Abstract]
Faries MB, Thompson JF, Cochran AJ, et al.: Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N Engl J Med 376 (23): 2211-2222, 2017. [PUBMED Abstract]
Leiter U, Stadler R, Mauch C, et al.: Complete lymph node dissection versus no dissection in patients with sentinel lymph node biopsy positive melanoma (DeCOG-SLT): a multicentre, randomised, phase 3 trial. Lancet Oncol 17 (6): 757-767, 2016. [PUBMED Abstract]
Luke JJ, Rutkowski P, Queirolo P, et al.: Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): a randomised, double-blind, phase 3 trial. Lancet 399 (10336): 1718-1729, 2022. [PUBMED Abstract]
Long GV, Luke JJ, Khattak MA, et al.: Pembrolizumab versus placebo as adjuvant therapy in resected stage IIB or IIC melanoma (KEYNOTE-716): distant metastasis-free survival results of a multicentre, double-blind, randomised, phase 3 trial. Lancet Oncol 23 (11): 1378-1388, 2022. [PUBMED Abstract]
Luke JJ, Ascierto PA, Khattak MA, et al.: Pembrolizumab Versus Placebo as Adjuvant Therapy in Resected Stage IIB or IIC Melanoma: Final Analysis of Distant Metastasis-Free Survival in the Phase III KEYNOTE-716 Study. J Clin Oncol 42 (14): 1619-1624, 2024. [PUBMED Abstract]
Treatment of Resectable Stage III Melanoma
Treatment Options for Resectable Stage III Melanoma
Treatment options for resectable stage III melanoma include the following:
The primary tumor may be treated with wide local excision with 1-cm to 2-cm margins, depending on tumor thickness and location.[1–7] Skin grafting may be necessary to close the resulting defect.
Lymph node management
Sentinel lymph node biopsy (SLNB)
Lymphatic mapping and SLNB can be considered to assess the presence of occult metastases in the regional lymph nodes of patients with primary tumors measuring at least 0.8 mm thick. These procedures may identify individuals who can avoid regional lymph node dissection and individuals who may benefit from adjuvant therapy.[3,8–12]
To ensure accurate identification of the sentinel lymph node, lymphatic mapping and removal of the sentinel lymph node are performed during the same operation as the wide excision of the primary melanoma.
Multiple studies have demonstrated the diagnostic accuracy of SLNB, with false-negative rates of 0% to 2%.[8,12–17] If micrometastatic melanoma is detected, active surveillance with ultrasound of the draining nodal basin is an acceptable treatment recommendation that has widely replaced complete lymph node dissection (CLND).[18,19] A complete regional lymphadenectomy can be considered in select populations.
Regional lymphadenectomy
In patients with microscopic melanoma in regional lymph nodes, immediate completion lymphadenectomy has widely been replaced by active observation, as long as close follow-up with nodal ultrasound surveillance can be achieved. Completion lymphadenectomy may still be considered on a case-by-case basis.
For patients who present with clinically detected (i.e., macroscopic) regional lymph node involvement in the absence of distant disease, therapeutic regional lymphadenectomy may be performed.
Neoadjuvant therapy
Resection of the bulk of the tumor and tumor-infiltrating lymphocytes may decrease the potential effect of programmed death-1 (PD-1) blockade in the adjuvant setting, leading investigators to study the use of neoadjuvant immunotherapy. Neoadjuvant pembrolizumab can be considered for patients with high-risk, node-positive disease or resectable metastatic disease. This treatment approach is based on the results of a randomized phase II trial in which 1 year of neoadjuvant and adjuvant pembrolizumab resulted in an improvement in event-free survival (EFS) compared with primary resection plus adjuvant pembrolizumab. The U.S. Food and Drug Administration (FDA) has not approved this regimen, pending the completion of a randomized phase III trial.
Immunotherapy
Checkpoint inhibitors
Pembrolizumab
Evidence (pembrolizumab):
An open-label phase II study (SWOG S1801 [NCT03698019]) conducted at 90 sites in the United States enrolled 313 patients with resectable macroscopic stage IIIB to stage IV melanoma. Patients were randomly assigned (1:1) to receive either three cycles of neoadjuvant pembrolizumab (200 mg intravenously [IV] every 3 weeks) followed by surgery and fifteen cycles of adjuvant pembrolizumab (n = 154 patients) or primary surgery followed by eighteen cycles of adjuvant pembrolizumab (n = 159 patients).[20] The primary end point was EFS in the intention-to-treat population. Events were defined as disease progression or toxic effects that precluded surgery; the inability to resect all gross disease; disease progression, surgical complications, or toxic effects of treatment that precluded the initiation of adjuvant therapy within 84 days after surgery; recurrence of melanoma after surgery; or death from any cause. Exclusion criteria included prior immunotherapy for melanoma, active autoimmune disease in patients who had received systemic treatment within 2 years before trial entry, uveal melanoma, and any history of brain metastasis.
The median duration of follow-up was 14.7 months in both groups. A total of 105 events occurred (38 in the neoadjuvant-adjuvant group and 67 in the adjuvant-only group). EFS was significantly longer in the neoadjuvant-adjuvant group than in the adjuvant-only group (P = .004). The EFS rate at 2 years was 72% (95% confidence interval [CI], 64%–80%) in the neoadjuvant-adjuvant group and 49% (95% CI, 41%–59%) in the adjuvant-only group.[20][Level of evidence B1]
At the time of data cutoff, 36 deaths (14 in the neoadjuvant-adjuvant group and 22 in the adjuvant-only group) were reported. This small number of deaths precluded a comparison of overall survival (OS). The benefit of neoadjuvant-adjuvant pembrolizumab was seen across all subgroups of patients including those with BRAF pathogenic variants.
Among the 152 patients in the neoadjuvant–adjuvant group who had received at least one dose of pembrolizumab, 11 (7%) had at least one grade 3 or 4 adverse event that was deemed by the investigators to be related to pembrolizumab. The incidence of adverse events of grade 3 or higher during adjuvant therapy was similar in the two groups (12% in the neoadjuvant-adjuvant group and 14% in the adjuvant-only group). No new toxic effects of pembrolizumab were observed in either trial group. No deaths attributed by the investigators to pembrolizumab occurred in either group.
Ipilimumab and nivolumab
Evidence (ipilimumab and nivolumab):
An open-label phase III study (NADINA [NCT04949113]) included patients with resectable macroscopic stage III melanoma. Patients were randomly assigned (1:1) to receive either two cycles of neoadjuvant ipilimumab plus nivolumab every 3 weeks, followed by lymph node dissection/resection of in-transit metastases or lymph node dissection followed by 12 cycles of adjuvant nivolumab every 4 weeks. A total of 423 eligible patients were randomly assigned (212 patients to the neoadjuvant group and 211 to the adjuvant-only group). Adjuvant therapy for patients assigned to the neoadjuvant group was dictated by pathological response. Patients who had a major pathological response (≤10% residual viable tumor) did not receive any adjuvant treatment. Patients who had a pathological partial response (11%–50%) or a pathological nonresponse (>50% residual tumor) received either adjuvant dabrafenib plus trametinib for 46 weeks or an additional 11 cycles of adjuvant nivolumab. The primary end point was EFS in the intention-to-treat population. Events were defined as the time from randomization to the occurrence of progression to unresectable melanoma before surgery, disease recurrence, or death due to melanoma or treatment.[21]
The median duration of follow-up was 10.6 months in both groups. A total of 100 events occurred (28 in the neoadjuvant-adjuvant group and 72 in the adjuvant-only group). EFS was significantly longer in the neoadjuvant group than in the adjuvant-only group. The 12-month EFS rate was 83.7% (95% CI, 73.8%–94.8%) in the neoadjuvant group and 57.2% (95% CI, 45.1%–72.7%) in the adjuvant-alone group.[21][Level of evidence B1] The benefit of neoadjuvant therapy was seen across all subgroups of patients, including those with BRAF pathogenic variants.
Among the 212 patients in the neoadjuvant group who received at least one dose of treatment, 47.2% had at least one grade 3 or 4 adverse event, compared with 34.1% in the adjuvant-only group. No new toxic effects of ipilimumab and nivolumab were observed in either trial group. One patient in the adjuvant group died of pneumonitis caused by treatment. No treatment-related deaths occurred in the neoadjuvant group.
Adjuvant therapy
Adjuvant therapeutic options are expanding for patients at high risk of recurrence after complete resection and include checkpoint inhibitors and combination signal transduction inhibitor therapy. Ipilimumab was the first checkpoint inhibitor to be approved by the FDA as adjuvant therapy. It has demonstrated improved OS when given at 10 mg/kg (ipi10), compared with placebo (EORTC 18071 [NCT00636168]).[22] However, ipi10 has significant toxicity at this dose. The North American Intergroup Trial E1609 (NCT01274338), designed with three treatment groups, compared ipi10 with ipilimumab at a lower dose of 3 mg/kg (ipi3) (approved for metastatic disease) and with high-dose interferon (HDI). Patients who received ipi3 had a significant improvement in OS compared with ipi10.[23] These data do not support HDI as an adjuvant treatment option for melanoma. As newer checkpoint inhibitors emerge, the role of ipi3 remains to be further defined.
Large randomized trials with the newer checkpoint inhibitors (nivolumab and pembrolizumab) and with combination signal transduction inhibitors (dabrafenib plus trametinib) showed a clinically significant impact on relapse-free survival (RFS). The CheckMate 238 (NCT02388906) trial compared nivolumab with ipi10 and found that nivolumab produced superior RFS and had a more tolerable safety profile.[24] Pembrolizumab produced superior RFS compared with placebo, with data on OS still maturing in the MK-3475-054/KEYNOTE-054 trial (NCT02362594).[25] Dabrafenib plus trametinib produced superior RFS compared with placebo, with data on OS still maturing in the COMBI-AD trial (NCT01682083).[26] Single-agent BRAF-inhibitor therapy with vemurafenib did not show improved RFS compared with placebo in the BRIM8 trial (NCT01667419).[27]
The benefit of immunotherapy with ipilimumab, nivolumab, and pembrolizumab has been seen regardless of programmed death-ligand 1 (PD-L1) expression or the presence of BRAF pathogenic variants. Combination signal transduction inhibitor therapy with dabrafenib plus trametinib is an additional option for patients with BRAF variants.
Patients should consider participating in clinical trials to identify treatments that will further extend RFS and OS with less toxicity and shorter treatment schedules.
Immunotherapy
Checkpoint inhibitors
Nivolumab
Evidence (nivolumab):
In a multinational double-blind trial (CheckMate 238 [NCT02388906]), patients with stage IIIB, IIIC, or IV melanoma who underwent complete resection were randomly assigned (1:1) to receive either nivolumab or ipilimumab.[24][Level of evidence B1] The primary end point was RFS and was defined as time from randomization until the date of the first recurrence, new primary melanoma, or death from any cause. Patients who were excluded included those with resection occurring more than 12 weeks before randomization, autoimmune disease, use of systemic glucocorticoids, previous systemic therapy for melanoma, and an Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) score higher than 1. Nivolumab (3 mg/kg) was given IV every 2 weeks and ipilimumab (10 mg/kg) was given every 3 weeks for four doses, then every 3 months for up to 1 year or until disease recurrence, along with corresponding placebo.
A total of 906 patients were randomly assigned: 453 patients to nivolumab and 453 patients to ipilimumab. Baseline characteristics were balanced in the two treatment groups. Approximately 81% of patients had stage III disease, 32% had ulcerated primary melanoma, 48% had macroscopic lymph node involvement, 62% had less than a 5% PD-L1 expression, and 42% had BRAF pathogenic variants.
The European Organisation for Research and Treatment of Cancer (EORTC) Independent Data Monitoring Committee stopped the study at the protocol-specified interim analysis, when all patients had a minimum follow-up of 18 months, at which time there were 360 events of RFS. The median RFS has not been reached in either treatment group. At 12 months, the RFS rate was 70.5% (95% CI, 66.1%–74.0%) for patients treated with nivolumab versus 60.8% (95% CI, 56.0%–65.2%) for patients treated with ipilimumab. Recurrence or death occurred in 34% (154 of 453) of patients treated with nivolumab versus 45.5% (206 of 453) of patients treated with ipilimumab (hazard ratio [HR] recurrence or death, 0.65; 97.56% CI, 0.51–0.83; P < .001). Subgroup analyses of RFS favored nivolumab regardless of PD-L1 expression or the presence of a BRAF V600 pathogenic variant.
Patients treated with nivolumab had fewer adverse events, including grade 3 to 4 serious adverse events and death. Adverse events led to treatment discontinuation in 9.7% of patients assigned to nivolumab and 42.6% of patients assigned to ipilimumab. Two treatment-related deaths occurred among patients treated with ipilimumab (e.g., marrow aplasia and colitis) and none occurred among the patients treated with nivolumab. The adverse event profile was like the type of checkpoint inhibitor toxicities seen in the metastatic setting, with immune-related events most commonly seen in the gastrointestinal system, hepatic system, and skin. Grade 3 or 4 adverse events occurred in 14% of patients treated with nivolumab and in 46% of patients treated with ipilimumab.
An updated analysis, after a minimum of 4 years of follow-up, showed an RFS rate of 51.7% (95% CI, 46.8%–56.3%) for patients who received nivolumab versus 41.2% (95% CI, 36.4%–45.9%) for patients who received ipilimumab (HR relapse or death, 0.71; 95% CI, 0.60–0.86; P = .0003). Median OS has not been reached in either group.[28]
Pembrolizumab
Evidence (pembrolizumab):
In a multinational double-blind trial (MK-3475-054/KEYNOTE-054 [NCT02362594]), patients with completely resected stage IIIA, IIIB, or IIIC melanoma were randomly assigned (1:1) to receive either pembrolizumab or placebo.[25][Level of evidence B1] The primary end point was RFS, defined as time from randomization until the date of first recurrence or death from any cause. If recurrence was documented, patients could cross over or repeat treatment with pembrolizumab. Pembrolizumab was given as an IV infusion of 200 mg every 3 weeks, for a total of 18 doses (approximately 1 year).
A total of 1,019 patients were randomly assigned: 514 to pembrolizumab and 505 to placebo. Baseline characteristics were balanced in the two treatment groups. Approximately 40% had ulcerated primary melanoma, 66% had macroscopic lymph node involvement, 84% had positive PD-L1 expression (melanoma score >2 by 22C3 antibody assay), and 44% had BRAF pathogenic variants.
The EORTC Independent Data Monitoring Committee reviewed the unblinded results at an amended interim analysis when 351 events (recurrences or deaths) had occurred. The results were positive, and the interim analysis of RFS became the final analysis.
At the amended interim analysis with a median follow-up of 15 months, the 12-month RFS rate was 75.4% (95% CI, 71.3%–78.9%) in the pembrolizumab group versus 61.0% (95% CI, 56.5%–65.1%) in the placebo group.
An update of the primary end point of RFS with a median follow-up 3.5 years showed an RFS rate of 59.8% (95% CI, 55.3%–64.1%) in the pembrolizumab group versus 41.4% (95% CI, 39.2%–48.8%) in the placebo group (HR, 0.59; 95% CI, 0.49–0.73).[29]
Pembrolizumab maintained its effect regardless of PD-L1 expression or BRAF pathogenic variant status.
Approximately 14% of patients discontinued pembrolizumab due to an adverse event. Grade 3, 4, or 5 adverse events considered to be related to pembrolizumab occurred in 15% of patients. There was one death due to treatment (myositis).
Ipilimumab
Evidence (ipilimumab):
The open-label, three-arm, North American Intergroup trial E1609 (NCT01274338) compared two doses of ipilimumab with HDI as adjuvant therapy in high-risk patients with melanoma.[23] A total of 1,670 patients with resected disease (defined by the American Joint Committee on Cancer, 7th edition, as stage IIIB, IIIC, M1a, or M1b) were randomly assigned (1:1:1) to ipi3 or ipi10 every 3 weeks for four doses (induction), followed by the same dose every 12 weeks for four doses (maintenance), or HDI 20 million units/m2 per day, 5 days per week for 4 weeks (induction), followed by 10 million units/m2 daily subcutaneously every other day, 3 days per week for 48 weeks (maintenance).[23][Level of evidence A1]
The trial was designed with two coprimary end points, RFS and OS, with a hierarchic analysis to evaluate ipi3 versus HDI followed by ipi10 versus HDI. The time to event was longer than anticipated and the design was amended for a final analysis at a data cutoff date giving a median follow-up time of 57.4 months (range, 0.03−86.6).
Ipi3 significantly improved OS compared with HDI (HR, 0.78; 95% repeated CI, 0.61−0.99; P = .044), but not RFS (HR, 0.85; 95% CI, 0.66−1.09; P = .065).
Ipi10 did not significantly improve OS or RFS compared with HDI (HR, 0.88; 95.6% CI, 0.69−1.12 and exploratory HR, 0.84; 99.4% CI, 0.65−1.09, respectively).
Salvage treatments were used in 69.7% of patients after ipi3, 51.6% of patients after ipi10, and 86.2% of patients after HDI.
Toxicity with ipi3 was lower than with ipi10; however, both had treatment-related discontinuations and death.
Treatment-related discontinuations occurred in 34.9% of the ipi3 group and in 54.1% of the ipi10 group.
Three possibly treatment-related deaths occurred in the ipi3 group, five in the ipi10 group, and two in the HDI group.
The study concluded that evidence no longer supports a role for HDI as adjuvant therapy for patients with high-risk melanoma. Further, ipi3 provides OS data superior to ipi10 compared with HDI. The role of ipilimumab as adjuvant monotherapy is unclear because the CheckMate 238 trial demonstrated that nivolumab was superior to ipi10 in improving RFS, with OS data still maturing.
In a multinational double-blind trial (EORTC 18071 [NCT00636168]), patients with stage III melanoma, who had complete resection, were randomly assigned (1:1) to receive either ipilimumab or placebo.[30][Level of evidence B1] Exclusion criteria comprised patients with lymph node metastasis larger than 1 mm, in-transit metastasis, resection occurring more than 12 weeks before randomization, autoimmune disease, previous or concurrent immunosuppressive therapy, previous systemic therapy for melanoma, and an ECOG PS score higher than 1. The ipilimumab dose was 10 mg/kg every 3 weeks for four doses, then every 3 months for up to 3 years. The primary end point was RFS, defined as recurrence or death (regardless of cause), whichever came first, as assessed by an independent review committee.
A total of 951 patients were enrolled (475 patients to the ipilimumab arm and 476 patients to the placebo arm). The median age was 51 years, and 94% of the patients had a PS of 0.
At a median follow-up of 2.7 years, there were 528 RFS events: 234 in the ipilimumab group (49%; 220 recurrences, 14 deaths) and 294 in the placebo group (62%; 289 recurrences, 5 deaths). Median RFS was 26 months for the ipilimumab group (95% CI, 19–39) versus 17 months for the placebo group (95% CI, 13–22). The HR was 0.75 (95% CI, 0.64–0.90; P < .002). The effect of ipilimumab was consistent across subgroups.
Ipilimumab was discontinued because of adverse events in 52% of the patients. Patients received ipilimumab for a median of four doses; 36% of patients in the ipilimumab group stayed on treatment for more than 6 months, and 26% stayed on treatment for more than 1 year. Five patients died from drug-related events: three secondary to colitis, one with myocarditis, and one of multiorgan failure with Guillain-Barré syndrome. The most common adverse events were gastrointestinal, hepatic, and endocrine related and included rash, fatigue, and headache.
An updated analysis was performed at a median follow-up of 5.3 years.[22]
The 5-year RFS rate was 40.8% in patients treated with ipilimumab and 30.3% in patients who received the placebo (HR recurrence or death, 0.76; 95% CI, 0.64–0.89; P < .001). The median RFS rate was 27.6% in patients treated with ipilimumab and 17.1% in patients who received the placebo.
The 5-year OS rate, a secondary end point, was 65.4% in patients treated with ipilimumab versus 54.4% in patients who received the placebo (HRdeath, 0.72; 95.1% CI, 0.58–0.88; P = .001).
Data from this trial (EORTC 18071), which tested high-dose ipilimumab at 10 mg/kg compared with placebo, served as the basis for the approval of ipilimumab in the adjuvant setting. However, the subsequent intergroup trial, E1609 (described above), demonstrated better outcomes with low-dose (3 mg/kg) ipilimumab, which is also the dose approved for metastatic disease.
Combination signal transduction inhibitors
Dabrafenib plus trametinib
Evidence (dabrafenib plus trametinib):
A multinational double-blind trial (COMBI-AD [NCT01682083]) included patients with stage IIIA, IIIB, or IIIC melanoma with BRAF V600E or V600K pathogenic variants who underwent completion lymphadenectomy. Patients were randomly assigned (1:1) to receive either dabrafenib plus trametinib or two matched placebo tablets.[26] The primary end point was RFS, defined as time from randomization until the date of first recurrence or death from any cause. Patients with resection occurring more than 12 weeks before random assignment and an ECOG PS score higher than 1 were excluded. Dabrafenib was given at a dose of 150 mg twice daily plus trametinib at a dose of 2 mg once daily (combination therapy) for 12 months in the absence of disease recurrence, unacceptable toxic effects, or death. A total of 870 patients were randomly assigned (438 patients to combination therapy and 432 patients to placebo). Baseline characteristics were balanced in the two treatment groups. Most patients (91%) had BRAF V600E pathogenic variants compared with 9% who had BRAF V600K variants. Most patients (92%) had an ECOG PS of 0.
At the data cutoff date for the primary analysis, the minimum follow-up was 2.5 years (median, 2.8 years), and all patients had completed trial treatment. Disease recurrence occurred in 163 of 438 patients (37%) who received combination therapy and in 247 of 432 patients (57%) who received placebo (HRrelapse or death, 0.47; 95% CI, 0.39–0.58; P < .001). Median RFS had not been reached in the combination arm (95% CI, 44.5–not reached) and was 16.6 months (95% CI, 12.7–22.1) in the placebo group.[26][Level of evidence B1]
In the combination therapy arm, 26% of the patients had an adverse event leading to the discontinuation of therapy, 38% required dose reduction, and 66% required dose interruption. In the placebo arm, 3% of the patients had an adverse event leading to discontinuation of therapy, 3% required dose reduction, and 15% required dose interruption. Serious adverse events occurred in 36% of the patients who received the combination therapy and 10% of the patients in the placebo group. One death, which resulted from pneumonia, was reported in the combination therapy arm.
A final analysis performed after a minimum of 8.33 years of follow-up showed superior RFS for patients who received the dabrafenib plus trametinib combination compared with patients who received placebo. Of patients who received the combination, 52% (95% CI, 47.4%–56.3%) were alive without relapse compared with 36% (95% CI, 30.9%–40.2%) of patients who received the placebo (HR, 0.52; 95% CI, 0.43–0.63). DMFS was also improved in patients who received the combination (60% vs. 46%; HR, 0.56; 95% CI, 0.44–0.71). An OS analysis showed improvement that was not statistically significant (HR, 0.80; 95% CI, 0.62–1.01).[31,32]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Veronesi U, Cascinelli N: Narrow excision (1-cm margin). A safe procedure for thin cutaneous melanoma. Arch Surg 126 (4): 438-41, 1991. [PUBMED Abstract]
Veronesi U, Cascinelli N, Adamus J, et al.: Thin stage I primary cutaneous malignant melanoma. Comparison of excision with margins of 1 or 3 cm. N Engl J Med 318 (18): 1159-62, 1988. [PUBMED Abstract]
Wagner JD, Gordon MS, Chuang TY, et al.: Current therapy of cutaneous melanoma. Plast Reconstr Surg 105 (5): 1774-99; quiz 1800-1, 2000. [PUBMED Abstract]
Cohn-Cedermark G, Rutqvist LE, Andersson R, et al.: Long term results of a randomized study by the Swedish Melanoma Study Group on 2-cm versus 5-cm resection margins for patients with cutaneous melanoma with a tumor thickness of 0.8-2.0 mm. Cancer 89 (7): 1495-501, 2000. [PUBMED Abstract]
Balch CM, Soong SJ, Smith T, et al.: Long-term results of a prospective surgical trial comparing 2 cm vs. 4 cm excision margins for 740 patients with 1-4 mm melanomas. Ann Surg Oncol 8 (2): 101-8, 2001. [PUBMED Abstract]
Heaton KM, Sussman JJ, Gershenwald JE, et al.: Surgical margins and prognostic factors in patients with thick (>4mm) primary melanoma. Ann Surg Oncol 5 (4): 322-8, 1998. [PUBMED Abstract]
Balch CM, Urist MM, Karakousis CP, et al.: Efficacy of 2-cm surgical margins for intermediate-thickness melanomas (1 to 4 mm). Results of a multi-institutional randomized surgical trial. Ann Surg 218 (3): 262-7; discussion 267-9, 1993. [PUBMED Abstract]
Shen P, Wanek LA, Morton DL: Is adjuvant radiotherapy necessary after positive lymph node dissection in head and neck melanomas? Ann Surg Oncol 7 (8): 554-9; discussion 560-1, 2000. [PUBMED Abstract]
Hochwald SN, Coit DG: Role of elective lymph node dissection in melanoma. Semin Surg Oncol 14 (4): 276-82, 1998. [PUBMED Abstract]
Cascinelli N, Morabito A, Santinami M, et al.: Immediate or delayed dissection of regional nodes in patients with melanoma of the trunk: a randomised trial. WHO Melanoma Programme. Lancet 351 (9105): 793-6, 1998. [PUBMED Abstract]
Koops HS, Vaglini M, Suciu S, et al.: Prophylactic isolated limb perfusion for localized, high-risk limb melanoma: results of a multicenter randomized phase III trial. European Organization for Research and Treatment of Cancer Malignant Melanoma Cooperative Group Protocol 18832, the World Health Organization Melanoma Program Trial 15, and the North American Perfusion Group Southwest Oncology Group-8593. J Clin Oncol 16 (9): 2906-12, 1998. [PUBMED Abstract]
Wong SL, Balch CM, Hurley P, et al.: Sentinel lymph node biopsy for melanoma: American Society of Clinical Oncology and Society of Surgical Oncology joint clinical practice guideline. J Clin Oncol 30 (23): 2912-8, 2012. [PUBMED Abstract]
Kirkwood JM, Strawderman MH, Ernstoff MS, et al.: Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14 (1): 7-17, 1996. [PUBMED Abstract]
Kirkwood JM, Ibrahim JG, Sondak VK, et al.: High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol 18 (12): 2444-58, 2000. [PUBMED Abstract]
Eggermont AM, Suciu S, Santinami M, et al.: Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet 372 (9633): 117-26, 2008. [PUBMED Abstract]
Hancock BW, Wheatley K, Harris S, et al.: Adjuvant interferon in high-risk melanoma: the AIM HIGH Study–United Kingdom Coordinating Committee on Cancer Research randomized study of adjuvant low-dose extended-duration interferon Alfa-2a in high-risk resected malignant melanoma. J Clin Oncol 22 (1): 53-61, 2004. [PUBMED Abstract]
Chapman PB, Hauschild A, Robert C, et al.: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364 (26): 2507-16, 2011. [PUBMED Abstract]
Faries MB, Thompson JF, Cochran AJ, et al.: Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N Engl J Med 376 (23): 2211-2222, 2017. [PUBMED Abstract]
Leiter U, Stadler R, Mauch C, et al.: Complete lymph node dissection versus no dissection in patients with sentinel lymph node biopsy positive melanoma (DeCOG-SLT): a multicentre, randomised, phase 3 trial. Lancet Oncol 17 (6): 757-767, 2016. [PUBMED Abstract]
Patel SP, Othus M, Chen Y, et al.: Neoadjuvant-Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N Engl J Med 388 (9): 813-823, 2023. [PUBMED Abstract]
Blank CU, Lucas MW, Scolyer RA, et al.: Neoadjuvant Nivolumab and Ipilimumab in Resectable Stage III Melanoma. N Engl J Med 391 (18): 1696-1708, 2024. [PUBMED Abstract]
Eggermont AM, Chiarion-Sileni V, Grob JJ, et al.: Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N Engl J Med 375 (19): 1845-1855, 2016. [PUBMED Abstract]
Tarhini AA, Lee SJ, Hodi FS, et al.: Phase III Study of Adjuvant Ipilimumab (3 or 10 mg/kg) Versus High-Dose Interferon Alfa-2b for Resected High-Risk Melanoma: North American Intergroup E1609. J Clin Oncol 38 (6): 567-575, 2020. [PUBMED Abstract]
Weber J, Mandala M, Del Vecchio M, et al.: Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N Engl J Med 377 (19): 1824-1835, 2017. [PUBMED Abstract]
Eggermont AMM, Blank CU, Mandala M, et al.: Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N Engl J Med 378 (19): 1789-1801, 2018. [PUBMED Abstract]
Long GV, Hauschild A, Santinami M, et al.: Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N Engl J Med 377 (19): 1813-1823, 2017. [PUBMED Abstract]
Maio M, Lewis K, Demidov L, et al.: Adjuvant vemurafenib in resected, BRAFV600 mutation-positive melanoma (BRIM8): a randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol 19 (4): 510-520, 2018. [PUBMED Abstract]
Ascierto PA, Del Vecchio M, Mandalá M, et al.: Adjuvant nivolumab versus ipilimumab in resected stage IIIB-C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol 21 (11): 1465-1477, 2020. [PUBMED Abstract]
Eggermont AMM, Blank CU, Mandalà M, et al.: Adjuvant pembrolizumab versus placebo in resected stage III melanoma (EORTC 1325-MG/KEYNOTE-054): distant metastasis-free survival results from a double-blind, randomised, controlled, phase 3 trial. Lancet Oncol 22 (5): 643-654, 2021. [PUBMED Abstract]
Eggermont AM, Chiarion-Sileni V, Grob JJ, et al.: Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol 16 (5): 522-30, 2015. [PUBMED Abstract]
Dummer R, Hauschild A, Santinami M, et al.: Five-Year Analysis of Adjuvant Dabrafenib plus Trametinib in Stage III Melanoma. N Engl J Med 383 (12): 1139-1148, 2020. [PUBMED Abstract]
Long GV, Hauschild A, Santinami M, et al.: Final Results for Adjuvant Dabrafenib plus Trametinib in Stage III Melanoma. N Engl J Med 391 (18): 1709-1720, 2024. [PUBMED Abstract]
Treatment of Unresectable Stage III, Stage IV, and Recurrent Melanoma
Treatment Options for Unresectable Stage III, Stage IV, and Recurrent Melanoma
Treatment options for unresectable stage III, stage IV, and recurrent melanoma include the following:
Complete surgical resection of all known disease versus best medical therapy (under clinical evaluation).
Isolated limb perfusion for unresectable extremity melanoma (under clinical evaluation).
Systemic therapy for unresectable disease (under clinical evaluation).
Two approaches—checkpoint inhibition and targeting the mitogen-activated protein kinase (MAPK) pathway—improved progression-free survival (PFS) and overall survival (OS) in randomized trials. Anti–PD-1 monotherapy (pembrolizumab or nivolumab) improved efficacy outcomes with better safety profiles when compared with treatment using single-agent anti–CTLA-4 (ipilimumab) or investigator choice of chemotherapy. The combination of anti–PD-1 and anti–CTLA-4 immunotherapies (nivolumab and ipilimumab) also prolongs PFS and OS compared with ipilimumab, but the combination is associated with significant toxicity. The efficacy seen with immunotherapy is independent of BRAF variant status.
Combinations of BRAF and MEK inhibitors have consistently shown superior efficacy compared with BRAF monotherapy. Improved PFS was seen when a PD-L1 inhibitor (atezolizumab) was added to the combination of a BRAF plus MEK inhibitor (vemurafenib plus cobimetinib); however, data on OS are immature. Further questions remain regarding triplet therapy, including how it compares with monotherapy checkpoint inhibition and if the concurrent administration is superior to sequential therapy (NCT02224781).
Because of the rapid development of new agents, combinations, and remaining questions, patients and their physicians are encouraged to consider a clinical trial for initial treatment and at the time of progression. Ongoing clinical trials address the following issues:
The value of sequencing therapies, such as immunotherapy and targeted therapy.
Optimal doses of combination immunotherapy to decrease toxicity and preserve efficacy.
How to select the patients who will benefit from combination immunotherapy versus monotherapy.
The role of PD-L1 expression as a biomarker for efficacy.
The role of maintenance therapy.
Immunotherapy
Checkpoint inhibitors
Anti–PD-1 and PD-L1 therapy
The PD-1 pathway is a key immunoinhibitory mediator of T-cell exhaustion. Blockade of this pathway can lead to T-cell activation, expansion, and enhanced effector functions. PD-1 has two ligands, PD-L1 and PD-L2. The U.S. Food and Drug Administration (FDA) has approved two anti–PD-1 antibodies, pembrolizumab and nivolumab, based on improved OS in randomized trials.
Pembrolizumab
Evidence (pembrolizumab):
Previously treated patients. One study included 173 patients with unresectable or metastatic melanoma with disease progression within 24 weeks of the last dose of ipilimumab. If the patient had a BRAF V600 pathogenic variant, previous treatment with a BRAF inhibitor was also required. Patients were randomly assigned to one of two doses of pembrolizumab—2 mg/kg or 10 mg/kg—every 3 weeks. The trial excluded patients with an autoimmune disease, a condition requiring immunosuppression, or a history of severe immune-related adverse events (irAEs) from treatment with ipilimumab.
The median age was 61 years; 60% were male; 67% had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) score of 0, and 33% had an ECOG PS of 1. Eighteen percent of patients had BRAF V600 pathogenic variants, 39% had an elevated lactate dehydrogenase (LDH) level, 64% had M1c disease, 9% had brain metastases, and 72% had undergone two or more therapies for advanced disease. The primary outcome measure was overall response rate according to Response Evaluation Criteria In Solid Tumors (RECIST, version 1.1) criteria as assessed by blinded independent central review.[1][Level of evidence B3]
The overall response rate was 26% (95% confidence interval [CI], -14 to 13; P = .96) for patients who received the 2 mg/kg dose, consisting of one complete response and 20 partial responses in 81 patients. Median follow-up was 8 months, and all patients had a minimum of 6 months of follow-up. Among the 21 patients with an objective response, 18 had ongoing responses, ranging from 1.4+ months to 8.5+ months.
Patients who received the 10 mg/kg dose had a similar response rate (26%), consisting of 20 responses in 76 patients. Responses were seen in patients with and without BRAF V600 pathogenic variants.
The approved dose was 2 mg/kg given as an intravenous (IV) infusion for 30 minutes every 3 weeks.
Pembrolizumab was discontinued because of adverse events in 7% of the patients treated with 2 mg/kg, with 3% considered drug-related adverse events by the investigators. The most common adverse events were the following (2 mg/kg vs. 10 mg/kg arms):
Fatigue (33% vs. 37%).
Pruritus (23% vs. 19%).
Rash (18% vs. 18%).
Other common adverse events included cough, nausea, decreased appetite, constipation, arthralgia, and diarrhea. The most frequent and serious adverse events that occurred in more than 2% of the 411 patients treated with pembrolizumab included renal failure, dyspnea, pneumonia, and cellulitis. Additional clinically significant irAEs included pneumonitis, colitis, hypophysitis, hyperthyroidism, hypothyroidism, nephritis, and hepatitis.
The FDA label provides recommendations for suspected irAEs, including withholding the drug and administering corticosteroids.
Previously untreated and treated patients. A multicenter international trial (KEYNOTE 006 [NCT01866319]) randomly assigned 834 patients with metastatic melanoma in a 1:1:1 ratio to receive pembrolizumab (10 mg/kg IV every 2 weeks or every 3 weeks) or four cycles of ipilimumab (3 mg/kg every 3 weeks).[2] Patients were stratified by ECOG PS (0 vs. 1), line of therapy (first-line vs. second-line), and PD-L1 expression (positive vs. negative). The primary end points were PFS and OS.[2][Level of evidence A1]
Approximately 66% of patients had received no previous systemic therapy for advanced melanoma. BRAF V600 pathogenic variants were present in 36% of patients and of these, approximately 50% had received previous BRAF inhibitor treatments. The study did not enroll patients with BRAF V600 pathogenic variants with high LDH levels and symptomatic or rapidly progressive disease who had not received anti-BRAF therapy, which could provide rapid clinical benefit. Approximately 80% of patients had PD-L1–positive tissue samples.
The final protocol-specified analysis of OS was conducted at a median follow-up of 23 months. Median OS was not reached in either pembrolizumab group; however, OS was 16.0 months for the ipilimumab group (hazard ratio [HR], 0.68; 95% CI, 0.53−0.87 for pembrolizumab every 2 weeks vs. ipilimumab; P = .0009 and 0.68; 95% CI, 0.53−0.86 for pembrolizumab every 3 weeks vs. ipilimumab; P = .0008). The 24-month survival rate was 55% in the groups who received pembrolizumab every 2 weeks and every 3 weeks compared with 43% in the ipilimumab group.[3]
Benefit was seen across all subgroups except for patients with PD-L1–negative tumors. However, since this subset was small (18% of patients) and the CI was wide, no definitive conclusions could be drawn from this study.
Nivolumab
Evidence (nivolumab):
Previously treated patients. Accelerated approval was based on a planned noncomparative interim analysis of the first 120 patients who received nivolumab with at least 6 months of follow-up from a multicenter, open-label trial (CheckMate 037 [NCT01721746]). In this trial, patients were randomly assigned (2:1) to receive either nivolumab (3 mg/kg every 2 weeks) or the investigator’s choice of chemotherapy (either dacarbazine 1,000 mg/m2 IV every 3 weeks or the combination of carboplatin [area under the curve 6] every 3 weeks plus paclitaxel 175 mg/m2 every 3 weeks).[FDA label][Level of evidence C3] Patients were required to have unresectable or metastatic melanoma that had progressed after treatment with ipilimumab. If the patient had a BRAF V600 pathogenic variant, previous treatment with a BRAF inhibitor was also required. The trial excluded patients with an autoimmune disease, a condition requiring immunosuppression, or a history of severe irAEs from treatment with ipilimumab.
The median age of patients was 58 years; 65% of patients were male; and ECOG PS was 0 in 58% of patients. BRAF V600 pathogenic variants were present in 22% of patients; 76% had M1c disease; 56% had elevated LDH levels; 18% had a history of brain metastases; and, 68% had previously received two or more systemic therapies for metastatic disease.
Objective response rate and OS were coprimary end points. The objective response rate was 32% (95% CI, 23%–41%), with four complete responses and 34 partial responses as assessed by RECIST 1.1 criteria and an independent central review. Among the 38 patients with responses, 33 (87%) had ongoing responses, with durations from 2.6+ to 10.0+ months.
Responses were seen in patients with and without BRAF V600 pathogenic variants.
Safety analysis is based on 268 patients. Nivolumab was discontinued because of adverse events in 9% of patients. Serious adverse events occurred in 41% of patients, and grade 3 and grade 4 adverse events occurred in 42% of patients. The most common adverse events were rash, cough, upper respiratory tract infection, and peripheral edema. Other important adverse events included ventricular arrhythmia, iridocyclitis, increased amylase and lipase, dizziness, and neuropathy.
The FDA label provides recommendations for suspected irAEs, including withholding the drug and administering corticosteroids.
Previously untreated patients. A double-blind multicenter trial (CheckMate 066 [NCT01721772]) included 418 patients with unresectable stage III or stage IV melanoma without BRAF pathogenic variants. Patients were randomly assigned (1:1) to receive either nivolumab (3 mg/kg every 2 weeks) and a dacarbazine-matched placebo (every 3 weeks) or dacarbazine (1,000 mg/m2 every 3 weeks with a nivolumab-matched placebo every 2 weeks). The primary end point was OS.[4][Level of evidence A1] The trial was conducted in 80 centers in Europe, Israel, Australia, Canada, and South America, which are countries where dacarbazine had been a standard first-line treatment in patients without BRAF pathogenic variants.
The Data and Monitoring Safety Board (DMSB) noted a potential difference in OS during safety review. In 2014, an abbreviated report from an unplanned interim–database lock was reviewed showing a significant difference in OS, in favor of nivolumab. The DMSB recommended that the study be unblinded and allow patients on dacarbazine to receive nivolumab. The intended sample size was approximately 410 patients; a total of 418 patients had been entered.
Results from the double-blind portion of the study before the crossover amendment showed that median OS was not reached in the nivolumab group and was 10.8 months (95% CI, 9.3–12.1) in the dacarbazine group. The 1-year OS rate was 72.9% (95% CI, 65.%–78.9%) in the nivolumab group and 42.1% (95% CI, 33.0%–50.9%) in the dacarbazine group. The HRdeath was 0.42 (99.79% CI, 0.25–0.73; P < .001).
The most common adverse events in the nivolumab group were fatigue (19.9%), pruritus (17%), nausea (16.5%), and diarrhea (16%). Adverse events led to treatment discontinuation in 6.8% of patients in the nivolumab group and 11.7% of patients in the dacarbazine group. Adverse events with potential immunological etiology that occurred included gastrointestinal, hepatic, pulmonary, renal, endocrine, and skin. However, most resolved with a delay in study treatment, glucocorticoid administration, or both per management guidelines for nivolumab. No deaths were attributed to drug-related adverse events in either group.
Change in dosing regimen for nivolumab in metastatic melanoma.
In a population pharmacokinetic response analysis and a dose/exposure-response analysis, the flat dose of 240 mg of nivolumab every 2 weeks was considered pharmacokinetically equivalent to the dosing regimen of 3 mg/kg. Clinical safety and efficacy at the two doses appeared similar across body weight and tumor types in melanoma, non-small cell lung cancer, and renal cell carcinoma.[5]
The dosing regimen approved by the FDA for monotherapy has changed from 3 mg/kg to 240 mg IV every 2 weeks until disease progression or intolerable toxicity. The dosing regimen of 1 mg/kg of IV nivolumab when combined with ipilimumab will remain unchanged until after therapy with ipilimumab is complete, when the regimen will change to a 240 mg dose every 2 weeks until disease progression or intolerable toxicity.
Ipilimumab is a human monoclonal antibody that binds to CTLA-4, thereby blocking its ability to downregulate T-cell activation, proliferation, and effector function.
Approved by the FDA in 2011, ipilimumab has demonstrated clinical benefit by prolonging OS in randomized trials. Two prospective, randomized, international trials, one each in previously untreated and treated patients, supported the use of ipilimumab.[6,7]
Evidence (ipilimumab):
Previously treated patients. A total of 676 patients with previously treated, unresectable stage III or stage IV disease, and who were HLA-A*0201-positive, were enrolled in a three-arm, multinational, randomized (3:1:1), double-blind, double-placebo trial. A total of 403 patients were randomly assigned to receive ipilimumab (3 mg/kg every 3 weeks for 4 doses) with glycoprotein 100 (gp100) peptide vaccine. One hundred thirty-seven patients received ipilimumab (3 mg/kg every 3 weeks for 4 doses), and 136 patients received the gp100 vaccine. Patients were stratified by baseline metastases and previous receipt or nonreceipt of IL-2 therapy. Eighty-two of the patients had metastases to the brain at baseline.[7][Level of evidence A1]
The median OS was 10 months in patients who received ipilimumab alone and 10.1 months in those who received ipilimumab with the gp100 vaccine, compared with 6.4 months for patients who received the vaccine alone (HR of ipilimumab alone vs. gp100 alone, 0.66; P < .003; HR of ipilimumab plus vaccine vs. gp100 alone, 0.68; P < .001).
An analysis at 1 year showed that 44% of patients who were treated with ipilimumab and 45% of those treated with ipilimumab and the vaccine were alive, compared with 25% of the patients who received the vaccine only.
Grade 3 or grade 4 irAEs occurred in 10% to 15% of patients treated with ipilimumab. These irAEs most often included diarrhea or colitis, and endocrine-related events (e.g., inflammation of the pituitary). These events required cessation of therapy and institution of anti-inflammatory agents such as corticosteroids or, in four cases, infliximab (an antitumor necrosis factor-alpha antibody).
There were 14 drug-related deaths (2.1%), and seven deaths were associated with irAEs.
Previously untreated patients. A multicenter, international trial included 502 patients who were untreated for metastatic disease (adjuvant treatment was allowed). Patients were randomly assigned (1:1) to receive either ipilimumab (10 mg/kg) plus dacarbazine (850 mg/m2) or placebo plus dacarbazine (850 mg/m2) at weeks 1, 4, 7, and 10 followed by dacarbazine alone every 3 weeks through week 22. Patients with stable disease or an objective response and no dose-limiting toxic effects received ipilimumab or placebo every 12 weeks thereafter as maintenance therapy. The primary end point was survival. Patients were stratified according to ECOG PS and metastatic stage. Approximately 70% of the patients had an ECOG PS of 0, and the remainder of the patients had an ECOG PS of 1. Approximately 55% of patients had stage M1c disease.[6][Level of evidence A1]
The median OS was 11.2 months (95% CI, 9.4–13.6) in the ipilimumab-dacarbazine group versus 9.1 months (95% CI, 7.8–10.5) in the placebo-dacarbazine group. In the ipilimumab-dacarbazine group, estimated survival rates were 47.3% at 1 year, 28.5% at 2 years, and 20.8% at 3 years (HRdeath, 0.72; P < .001). In the placebo-dacarbazine group, the survival rates were 36.3% at 1 year, 17.9% at 2 years, and 12.2% at 3 years.
The most common study-drug–related adverse events were those classified as immune related. Grade 3 or grade 4 irAEs were seen in 38.1% of patients treated with ipilimumab plus dacarbazine versus 4.4% of patients treated with placebo plus dacarbazine. The most common events were hepatitis and enterocolitis.
No drug-related deaths occurred.
Clinicians and patients should be aware that immune-mediated adverse reactions may be severe or fatal. Early identification and treatment are necessary, including potential administration of systemic glucocorticoids or other immunosuppressants according to the immune-mediated adverse reaction management guide provided by the manufacturer.[8]
High-dose IL-2
The FDA approved IL-2 in 1998 because of durable complete responses in eight phase I and II studies. Phase III trials comparing high-dose IL-2 to other re-treatments, providing an assessment of relative impact on OS, have not been conducted.
Evidence (high-dose IL-2):
Based on a pooled analysis of 270 patients from eight single- and multi-institutional trials in 22 institutions conducted between 1985 and 1993, results included:
High-dose IL-2 demonstrated a complete response rate of 6% to 7%.[9]
With a median follow-up time for surviving patients of at least 7 years, the median duration of complete responses was not reached but was at least 59 months.[10]
Strategies to improve this therapy are an active area of investigation.
Dual checkpoint inhibition
T cells coexpress several receptors that inhibit T-cell function. Preclinical data and early clinical data suggest that co-blockade of two inhibitory receptors may be more effective than blockade of either alone.[11] This led to a phase III trial (NCT01844505) comparing each single agent with the combination of ipilimumab and nivolumab, and another phase III trial comparing nivolumab to the combination of nivolumab and relatlimab (NCT03470922). The FDA has approved both the ipilimumab-nivolumab and the nivolumab-relatlimab combinations.
CTLA-4 inhibitor plus PD-1 inhibitor
Evidence (ipilimumab plus nivolumab):
Previously untreated patients. In an international double-blind trial (CheckMate 067), 945 previously untreated patients with unresectable stage III or IV melanoma were randomly assigned in a 1:1:1 ratio to receive one of the following regimens:
Arm 1: nivolumab (3 mg/kg every 2 weeks) plus placebo;
Arm 2: nivolumab (1 mg/kg every 3 weeks) plus ipilimumab (3 mg/kg every 3 weeks for 4 doses) followed by nivolumab (3 mg/kg every 2 weeks for cycle 3 and beyond); or
Arm 3: ipilimumab (3 mg/kg every 3 weeks for 4 doses) plus placebo.
PFS and OS were coprimary end points. The study was powered to compare the combination of nivolumab plus ipilimumab with ipilimumab monotherapy, and nivolumab monotherapy with ipilimumab monotherapy. The study was not powered to compare combination ipilimumab plus nivolumab with nivolumab.
Patients were stratified according to tumor PD-L1 status assessed in a central laboratory by immunohistochemical testing (positive vs. negative or indeterminate), BRAF pathogenic variant status (V600 variant−positive vs. wild-type), and American Joint Committee on Cancer stage. Characteristics at baseline were as follows: 74% of patients had an ECOG PS of 0; 36% had elevated LDH levels; 31.5% had BRAF pathogenic variants; and 58% had M1c disease. A minority of patients (23.6%) had PD-L1–positive tumors.[12][Level of evidence A1]
The prospectively defined coprimary analysis of PFS occurred after all patients had at least 9 months of follow-up. Treatment with nivolumab alone or in combination with ipilimumab resulted in significantly longer PFS than with ipilimumab alone. Results were consistent across the prespecified stratification factors. Median PFS was 6.9 months (95% CI, 4.3–9.5) with nivolumab, 11.5 months (95% CI, 8.9–16.7) with nivolumab plus ipilimumab, and 2.9 months (95% CI, 2.8–3.4) with ipilimumab.
The prospectively specified coprimary analysis of OS was to occur at 28 months. With 467 deaths, the OS rate at this time point was 59% in the nivolumab group, 64% in the combination group, and 45% in the ipilimumab group (HRdeath for the combination vs. ipilimumab, 0.55 [98% CI, 0.42–0.72; P< .001]; HRdeath with nivolumab vs. ipilimumab 0.63 [98% CI, 0.48–0.81; P< .001]).[13]
In a descriptive analysis with a minimum follow-up of 36 months, the following data were found:
OS rates were 52% in the nivolumab group, 58% in the combination group, and 34% in the ipilimumab group.
The median OS was not reached in the combination arm (95% CI, 38.2 months–not reached). Median OS in the single-agent nivolumab and ipilimumab groups were 37.6 months (95% CI, 29.1–not reached) and 19.9 months (95% CI, 16.9–24.6), respectively.
For the combination versus ipilimumab, the HRdeath was 0.55 (99.5% CI, 0.45–0.69; P < .001); for nivolumab versus ipilimumab, the HR was 0.65 (99.5% CI, 0.53–0.80; P < .001).
Adverse events were highest in the combination arm and need to be monitored carefully. Grades 3 to 4 treatment-related adverse events occurred in 16.3% of patients in the nivolumab group, 27.3% of patients in the ipilimumab group, and 55% of patients in the combination group. The most frequent reason for treatment discontinuation was disease progression in the two monotherapy arms—49% with nivolumab and 65% with ipilimumab. The most frequent reason for discontinuation in the combination group was toxicity (38%).
Four therapy-related deaths were reported, which were attributed to neutropenia (nivolumab group), colon perforation (ipilimumab group), liver necrosis, and autoimmune myocarditis (combination ipilimumab and nivolumab).
Analyses of PD-L1 expression level associated with OS at 3 years showed that expression levels alone are not a reliable predictor of OS.
In a final analysis with a minimum follow-up of 10 years, the following data were reported:
The 10-year OS rate was 34% in the combination group, 26% in the nivolumab group, and 16% in the ipilimumab group.
The median OS was 71.9 months (95% CI, 51.6–not reached) in the combination group, 36.9 months (95% CI, 30.7–46.6) in the nivolumab group, and 19.9 months (95% CI, 16.8–24.6) in the ipilimumab group.
HRdeath, compared with the ipilimumab group, was 0.53 (95% CI, 0.44–0.65) for the combination group and 0.70 (95% CI, 0.59–0.84) for the nivolumab group.[14]
Melanoma metastatic to the brain. Patients with at least one measurable, nonirradiated brain metastasis were eligible for treatment with systemic dual immunotherapy in an open-label, multicenter phase II trial (CheckMate 204 [NCT02320058]).[15][Level of evidence C3] Eligibility required no need for immediate intervention, an absence of neurological signs or symptoms, and no glucocorticoids within 14 days of study treatment. Patients may have received previous stereotactic radiosurgery or excision of up to three brain metastases. Positive PD-L1 expression was not required.
Treatment consisted of nivolumab (1 mg/kg) plus ipilimumab (3 mg/kg) every 3 weeks for up to 4 doses, followed by nivolumab (3 mg/kg) every 2 weeks until progression or unacceptable toxicity.
The primary end point was rate of intracranial clinical benefit assessed by the investigator per RECIST criteria and defined as the percentage of patients with a complete response, partial response, or stable disease for at least 6 months. A total of 28 sites in the United States enrolled 101 patients, 94 of whom had a minimum follow-up of 6 months; the data on that population are reported below.
Clinical benefit (in the brain) was seen in 57% of patients (95% CI, 47%–68%); 24 patients (26%) had a complete response, 28 patients (30%) had a partial response, and 2 patients (2%) had stable disease that lasted for 6 months or longer. Similar rates of objective response (50%) were seen in patients with extracranial lesions, although fewer patients had a complete response (7%).
A subgroup analysis indicated responses in both PD-L1–positive and PD-L1–negative patients (baseline status not known in 20/94 patients).
The median follow-up of the 94 patients was 14 months. Median time to intracranial response was 2.3 months (range, 1.1−10.8), and time to extracranial response was 2.1 months (range, 1.1−15.0).
The most common treatment-related adverse event of any grade in the nervous system was headache (21 patients [22%]), with 3 patients (3%) having headaches of grade 3 or 4. Other treatment-related neurological adverse events of grade 3 or 4 were brain edema (2 patients [2%]), intracranial hemorrhage (1 patient [1%]), peripheral motor neuropathy (1 patient [1%]), and syncope (1 patient [1%]). Each of these adverse events led to treatment discontinuation, and the one reported case of peripheral motor neuropathy was irreversible. The investigator determined one death to be related to the study treatment (grade 5 immune-related myocarditis).
Progression was documented in 33 patients (35%); 17 patients (18%) had intracranial progression only, 4 patients (4%) had extracranial progression only, and 12 patients (13%) had progression in both intracranial and extracranial sites.
LAG-3 inhibitor plus PD-1 inhibitor
Evidence (relatlimab plus nivolumab):
A multinational, phase II/III, double-blind trial (RELATIVITY-047 [NCT03470922]) included 714 patients with previously untreated, histologically confirmed, unresectable stage III or IV melanoma. Patients were randomly assigned in a 1:1 ratio to receive either 160 mg of relatlimab and 480 mg of nivolumab in a fixed-dose combination (n = 355) or 480 mg of nivolumab (n = 359). The characteristics of the patients at baseline were well balanced between the treatment groups. Both therapies were given in a single 60-minute IV infusion every 4 weeks. The primary end point was PFS assessed according to RECIST, version 1.1. Secondary end points included OS and objective response. Patients who had received previous adjuvant or neoadjuvant therapies containing a PD-1, CTLA-4, BRAF, or MEK inhibitor (or a combination of BRAF and MEK inhibitors) were eligible if the therapy was completed at least 6 months before the date of recurrence. Key exclusion criteria were uveal melanoma and active, untreated brain or leptomeningeal metastases.[16]
The median PFS was 10.1 months (95% CI, 6.4–15.7) in the relatlimab-nivolumab group and 4.6 months (95% CI, 3.4–5.6) in the nivolumab-alone group (HRprogression or death, 0.75; 95% CI, 0.62–0.92; P = .006). The 12-month PFS rate was 47.7% (95% CI, 41.8%–53.2%) in the relatlimab-nivolumab group and 36.0% (95% CI, 30.5%–41.6%) in the nivolumab-alone group.[16][Level of evidence B1]
Grade 3 or 4 treatment-related adverse events occurred in 18.9% of the patients in the relatlimab-nivolumab group and 9.7% of patients in the nivolumab-alone group. Treatment-related adverse events led to treatment discontinuation in 14.6% of patients in the relatlimab-nivolumab group and 6.7% of patients in the nivolumab-alone group. The most common categories of immune-mediated adverse events that occurred in the relatlimab-nivolumab group were hypothyroidism or thyroiditis (18.0%), rash (9.3%), and diarrhea or colitis (6.8%). Myocarditis occurred in 1.7% of the patients in the relatlimab-nivolumab group and 0.6% of patients in the nivolumab-alone group.
The relatlimab-nivolumab combination was beneficial compared with nivolumab alone, regardless of LAG-3 expression or BRAF pathogenic variant status. OS and response rate data have not been reported.
Signal transduction inhibitors
Studies indicate that both BRAF and MEK inhibitors, as single agents and in combination, can significantly impact the natural history of melanoma, although they do not appear to provide a cure.
BRAF inhibitors
Treatment with BRAF inhibitors is discouraged for patients with wild-type BRAF melanoma because data from preclinical models have demonstrated that BRAF inhibitors can enhance rather than downregulate the MAPK pathway in tumor cells with wild-type BRAF and upstream RAS pathogenic variants.[17–20]
Vemurafenib
Vemurafenib is an orally available, small-molecule, selective BRAF kinase inhibitor that was approved by the FDA in 2011 for patients with unresectable or metastatic melanoma and BRAF V600E pathogenic variants.
Evidence (vemurafenib):
Previously untreated patients. The approval of vemurafenib was supported by an international multicenter trial (BRIM-3 [NCT01006980]) that screened 2,107 patients with previously untreated stage IIIC or IV melanoma for BRAF V600 pathogenic variants and identified 675 patients via the cobas 4800 BRAF V600 Mutation Test.[21] Patients were randomly assigned to receive either vemurafenib (960 mg by mouth twice a day) or dacarbazine (1,000 mg/m2 IV every 3 weeks). Coprimary end points were rates of OS and PFS. At the planned interim analysis, the DMSB determined that both the OS and PFS end points had met the prespecified criteria for statistical significance in favor of vemurafenib and recommended that patients in the dacarbazine group be allowed to cross over to receive vemurafenib.[21][Levels of evidence A1 and B1]
A total of 675 patients were evaluated for OS. Although the median survival had not yet been reached for vemurafenib and the data were immature for reliable Kaplan-Meier estimates of survival curves, the OS in the vemurafenib arm was clearly superior to that in the dacarbazine arm.
The HRdeath in the vemurafenib group was 0.37 (95% CI, 0.26–0.55; P < .001). The survival benefit in the vemurafenib group was observed in each prespecified subgroup, for example, age, sex, ECOG PS, tumor stage, LDH level, and geographical region.
The HR for tumor progression was 0.26 (95% CI, 0.20–0.33; P < .001) in the vemurafenib arm. The estimated median PFS was 5.3 months in the vemurafenib arm versus 1.6 months in the dacarbazine arm.
Twenty patients had non-BRAF V600E pathogenic variants: 19 with BRAF V600K and 1 with BRAF V600D. Four patients with BRAF V600K pathogenic variants had a response to vemurafenib.
Adverse events required dose modification or interruption in 38% of patients who received vemurafenib and 16% of those who received dacarbazine. The most common adverse events with vemurafenib were cutaneous events, arthralgia, and fatigue. Cutaneous squamous cell carcinoma (SCC), keratoacanthoma, or both developed in 18% of patients and were treated by simple excision. The most common adverse events with dacarbazine were fatigue, nausea, vomiting, and neutropenia. For more information, visit Fatigue and Nausea and Vomiting Related to Cancer Treatment.
Previously treated patients. A multicenter phase II trial included 132 patients with BRAF V600E or BRAF V600K pathogenic variants. Patients received vemurafenib (960 mg by mouth twice a day). Of the enrolled patients, 61% had stage M1c disease, and 49% had an elevated LDH level. All patients had received one or more previous therapies for advanced disease. Median follow-up was 12.9 months.[22][Level of evidence C3]
An independent review committee (IRC) reported a response rate of 53% (95% CI, 44%–62%), with eight patients (6%) achieving a complete response.
Median duration of response per IRC assessment was 6.7 months (95% CI, 5.6–8.6). Most responses were evident at the first radiological assessment at 6 weeks. However, some patients did not respond until after receiving therapy for more than 6 months.
Dabrafenib
Dabrafenib is an orally available, small-molecule, selective BRAF inhibitor that was approved by the FDA in 2013. It is used for the treatment of patients with unresectable or metastatic melanoma and BRAF V600E pathogenic variants as detected by an FDA-approved test. Dabrafenib and other BRAF inhibitors are not recommended for the treatment of BRAF wild-type melanomas, as in vitro experiments suggest there may be a paradoxical stimulation of MAPK signaling resulting in tumor promotion.
Evidence (dabrafenib):
An international multicenter trial (BREAK-3 [NCT01227889]) compared dabrafenib with dacarbazine. A total of 250 patients with unresectable stage III or IV melanoma and BRAF V600E pathogenic variants were randomly assigned in a 3:1 ratio to receive dabrafenib (150 mg by mouth daily) or dacarbazine (1,000 mg/m2 IV every 3 weeks). IL-2 was allowed as a previous treatment for advanced disease. The primary end point was PFS; patients could cross over at the time of progressive disease after confirmation by a blinded IRC.[23][Level of evidence B1]
With 126 events, the HR for PFS was 0.30 (95% CI, 0.18–0.51; P < .0001). The estimated median PFS was 5.1 months for dabrafenib versus 2.7 months for dacarbazine. OS data are limited by the median duration of follow-up and crossover. The partial response rate was 47% and the complete response rate was 3% in patients who received dabrafenib, compared with a partial response rate of 5% and a complete response rate of 2% for those who received dacarbazine.
The most frequent adverse events in patients treated with dabrafenib were cutaneous findings (i.e., hyperkeratosis, papillomas, palmar-plantar erythrodysesthesia), pyrexia, fatigue, headache, and arthralgia. Cutaneous SCC or keratoacanthoma occurred in 12 patients, basal cell carcinoma occurred in four patients, mycosis fungoides occurred in one patient, and new melanoma occurred in two patients.
MEK inhibitors
Trametinib
Trametinib is an orally available, small-molecule, selective inhibitor of MEK1 and MEK2. BRAF activates MEK1 and MEK2 proteins, which in turn, activate MAPK. Preclinical data suggest that MEK inhibitors can restrain growth and induce cell death of some BRAF-altered human melanoma tumors.
In 2013, the FDA approved trametinib for patients with unresectable or metastatic melanoma with BRAF V600E or V600K pathogenic variants, as determined by an FDA-approved test.
Evidence (trametinib):
A total of 1,022 patients were screened for BRAF pathogenic variants, resulting in 322 eligible patients (281 with BRAF V600E, 40 with BRAF V600K, and one with both variants).[24] One previous treatment (biological or chemotherapy) was allowed; however, no previous treatment with a BRAF or MEK inhibitor was permitted. Patients were randomly assigned in a 2:1 ratio to receive either trametinib (2 mg every day) or IV chemotherapy (either dacarbazine 1,000 mg/m2 every 3 weeks or paclitaxel 175 mg/m2 every 3 weeks). Crossover for patients randomly assigned to chemotherapy was allowed. The primary end point was PFS.
The investigator-assessed PFS was 4.8 months in patients who received trametinib and 1.5 months in patients who received chemotherapy (HR for PFS or death, 0.45; 95% CI, 0.33–0.63; P < .001). A radiology review blinded-to-treatment arm resulted in similar outcomes. Median OS has not been reached.
Adverse events leading to dose interruptions occurred in 35% of patients in the trametinib group and 22% of those in the chemotherapy group. Adverse events leading to dose reductions occurred in 27% of patients who received trametinib and in 10% of those who received chemotherapy.
The most common adverse events included rash, diarrhea, nausea, vomiting, fatigue, peripheral edema, alopecia, hypertension, and constipation. Cardiomyopathy (7%), interstitial lung disease (2.4%), central serous retinopathy (<1%), and retinal-vein occlusion (<1%) are uncommon but serious adverse events associated with trametinib. On-study cutaneous SCCs were not observed. For more information, visit Fatigue and Nausea and Vomiting Related to Cancer Treatment.
Cobimetinib
Cobimetinib is a small-molecule, selective MEK inhibitor that the FDA approved in 2015 for use in combination with the BRAF inhibitor vemurafenib. For more information, visit the Combination therapy with signal transduction inhibitors section.
KIT inhibitors
Early data suggest that mucosal or acral melanomas with activating pathogenic variants or amplifications in KIT may be sensitive to a variety of c-KIT inhibitors.[25–27] Phase II and phase III trials are available for patients with unresectable stage III or stage IV melanoma and KIT pathogenic variants.
Combination therapy with signal transduction inhibitors
Results from phase III trials comparing three different combinations of BRAF-MEK inhibitors with BRAF inhibitor monotherapy have consistently shown that combination therapy is superior to BRAF monotherapy.
Secondary resistance to BRAF inhibitor monotherapy in patients with BRAF V600 pathogenic variants may be associated with reactivation of the MAPK pathway. Therefore, combinations of signal transduction inhibitors that block different sites in the same pathway or sites in multiple pathways are an active area of research.
BRAF inhibitor plus MEK inhibitors
Dabrafenib plus trametinib
Evidence (dabrafenib plus trametinib):
Previously untreated. An international, double-blind, phase III trial (COMBI-d [NCT01584648]) without crossover included 423 previously untreated patients with unresectable stage IIIC or stage IV melanoma and BRAF V600E or V600K pathogenic variants. Patients were randomly assigned to receive either the combination of dabrafenib (150 mg by mouth twice a day) plus trametinib (2 mg by mouth every day) or dabrafenib plus placebo. The primary end point was investigator-assessed PFS. The protocol included a prespecified interim analysis for OS at the time of analysis of the primary end point. Patients were stratified by baseline LDH levels and BRAF genotype.[28][Level of evidence B1]
Median PFS was 9.3 months for the combination versus 8.8 months for dabrafenib plus placebo. The HRdeath or progression was 0.75 (95% CI, 0.57–0.99; P = .03). Updated data at the time of final analysis of OS revealed a median PFS of 11.0 months for the combination versus 8.8 months for dabrafenib plus placebo. The HRPFS or death was 0.67 (95% CI, 0.53–0.84; P = .0004; unadjusted for multiple testing).[29]
A prespecified final analysis of OS was conducted at 70% of events. Median OS was 25.1 months in the dabrafenib-plus-trametinib group (66% of events) versus 18.7 months in the dabrafenib-plus-placebo group (76% of events). The HR was 0.71 (95% CI, 0.55–0.92; P = 0.01).
Permanent discontinuations of study drugs were reported in 9% of patients who received the combination and in 5% of patients treated with dabrafenib only.
The incidence of grade 3 to grade 4 adverse events was similar between the groups (35% with the combination and 37% with dabrafenib only). Pyrexia occurred more frequently with the combination and was treated with immediate temporary cessation of the study drug in either group; prophylactic glucocorticoids may prevent recurring episodes. Hyperproliferative cutaneous events, including cutaneous SCCs, were considered related to paradoxical activation of the MAPK pathway and occurred less frequently with the addition of the MEK inhibitor. Rare, but serious, adverse events included decreased ejection fraction and chorioretinopathy.
Previously untreated. An international, open-label, phase III trial (COMBI-v [NCT01597908]) included 704 previously untreated patients with metastatic melanoma and BRAF V600 pathogenic variants. Patients were randomly assigned to receive standard doses of either the combination of dabrafenib plus trametinib or vemurafenib as first-line therapy. The primary end point was OS.[30][Level of evidence A1]
An interim analysis for OS was planned when 202 of the final 288 events occurred. Per protocol, the DMSB used adjusted efficacy boundaries for actual events (222) (2-sided P < .0214 for efficacy and P > .2210 for futility). The DMSB recommended stopping for efficacy, and the interim analysis is considered to be the final analysis of OS. A protocol amendment was issued to allow crossover to the combination therapy arm.
A total of 100 patients (28%) in the combination arm and 122 (35%) in the vemurafenib group had died (HR, 0.69; 95% CI, 0.53–0.89; P = .005). Median OS for patients treated with vemurafenib was 17.2 months; the median has not been reached in the combination therapy arm.
Previously untreated. A pooled analysis of the 563 patients who were randomly assigned to receive dabrafenib plus trametinib in the COMBI-d (double-blinded) and COMBI-v (open label) trials (described above) provides estimated 5-year outcomes.[31] After the study, 53% of patients received subsequent treatment; two-thirds of these patients received immunotherapy.
Sixty-three percent of patients died (64% in COMBI-d and 61% in COMBI-v) after a median follow-up of 22 months (range, 0−76 months).
The estimated 5-year OS rate was 34% (95% CI, 30%−38%), and the investigator-assessed PFS rate was 19% (95% CI, 15%−22%).
A complete response, which occurred in 109 patients (19%), was associated with an OS rate of 71% (95% CI, 62%−79%) at 5 years.
Vemurafenib plus cobimetinib
Evidence (vemurafenib plus cobimetinib):
Previously untreated. An international phase III trial included 495 patients with previously untreated, unresectable, stage IIIC or stage IV melanoma and BRAF V600 pathogenic variants. Patients were randomly assigned to receive either the combination of vemurafenib (960 mg by mouth every day) and cobimetinib (60 mg by mouth every day for 21 days followed by a 7-day rest period) or vemurafenib plus placebo. The primary end point was investigator-assessed PFS. Crossover at time of PFS was not allowed. Patients were stratified by stage and geographic region. Two interim analyses of OS were prespecified, with the first specified at the time of analysis of the primary end point.[32][Level of evidence B1]
The median PFS was 9.9 months in patients who received the combination versus 6.2 months in patients treated with vemurafenib plus placebo. The HRprogression or death was 0.51 (95% CI, 0.39–0.68; P = .001).
The first interim analysis of OS is immature because of the few events in both arms; therefore, median survival was not reached in either study group.
Rate of withdrawal of therapy caused by adverse events was similar between the groups (13% for patients treated with the combination and 12% for patients treated with vemurafenib only). Six deaths were attributed to adverse events in the combination group, and three deaths were attributed to adverse events in the vemurafenib-only group.
The incidence of grade 3 to grade 4 adverse events was similar between the groups (62% in patients treated with the combination and 58% in patients treated with vemurafenib alone). Rare, but serious, adverse events included chorioretinopathy, retinal detachment, decreased ejection fraction, and QT prolongation. Hyperproliferative cutaneous events, including cutaneous SCC, were considered to be related to paradoxical activation of the MAPK pathway and occurred less frequently with the addition of the MEK inhibitor.
Encorafenib plus binimetinib
Encorafenib is a small-molecule BRAF inhibitor, and binimetinib is a small-molecule MEK inhibitor. The combination of these two agents is approved for the treatment of unresectable or metastatic melanoma with BRAF V600E or V600K pathogenic variants, as detected by an FDA-approved test. The combination has demonstrated improved PFS and OS compared with vemurafenib. However, neither is approved as single-agent therapy.
Evidence (encorafenib plus binimetinib):
Previously untreated or progression on or after first-line immunotherapy. An international, open-label, phase III trial (COLUMBUS [NCT01909453]) included 577 patients with stage IIIB, IIIC, or IV melanoma and BRAF V600 pathogenic variants. Patients were randomly assigned in a 1:1:1 ratio to receive encorafenib (450 mg every day) plus binimetinib (45 mg twice a day), encorafenib monotherapy (300 mg every day), or vemurafenib monotherapy (960 mg twice a day).[33,34] The primary end point was PFS for the combination versus vemurafenib alone as assessed by a blinded IRC with a secondary end point of OS.
Approximately 5% of patients had received previous checkpoint inhibitor therapy.
With a median follow-up of 16.6 months, the median PFS was 14.9 months (95% CI, 11.0–18.5) with the combination versus 7.3 months (95% CI, 5.6–8.2) with vemurafenib alone. The HRprogression or death was 0.54 (95% CI, 0.41–0.71; 2-sided P < .0001).
With a median follow-up of 36.8 months for the secondary end point of OS, the median OS was 33.6 months (95% CI, 24.4–39.2) for patients in the combination arm and 16.9 months (95% CI, 14.0–24.5) for patients treated with vemurafenib alone (HR, 0.61; 95% CI, 0.47–0.79; P < .0001). Subsequent treatments after discontinuation of the study drug were received by 42% of patients in the combination group and 62% of patients in the vemurafenib-alone group.[33,34][Level of evidence A1]
The incidence of grade 3 to 4 adverse events was 58% with combination therapy and 63% with vemurafenib alone. Serious adverse events occurred in 34% of the combination group and 37% of the vemurafenib-alone group. The most common adverse events in the combination group included gastrointestinal symptoms and elevation of gamma-glutamyl transferase (GGT), elevation of creatine phosphokinase (CPK), left ventricular dysfunction (8%), and serous retinopathy (20%), mostly grade 1 to 2 (monitoring guidelines are provided in the drug label). Patients who received vemurafenib had more pyrexia and cutaneous toxicities. Study drug discontinuations from adverse events occurred in 15% of patients in the combination group and 17% of patients in the vemurafenib-alone group. No deaths were related to treatment; however, one death from suicide occurred in the combination arm.
Combination signal transduction inhibitor therapy plus PD-L1 inhibitor
Cobimetinib and vemurafenib plus atezolizumab
Evidence (cobimetinib and vemurafenib plus atezolizumab):
A double-blind, placebo-controlled, multicenter trial (IMspire150 [NCT02908672]) included 514 patients with unresectable stage IIIC or metastatic melanoma and BRAF V600 pathogenic variants. Patients were randomly assigned (1:1) to receive first-line therapy with cobimetinib plus vemurafenib with either atezolizumab or placebo.[35] Eligibility criteria included ECOG PS scores of 0 to 1, measurable disease, and no previous systemic treatment for metastatic melanoma. Patients with untreated or actively progressing brain metastases or a history of serious autoimmune disease were excluded. Previous adjuvant therapy was allowed (14% of patients).
After all patients in both arms received a 28-day cycle of cobimetinib and vemurafenib, patients received atezolizumab (840 mg IV every 2 weeks) or placebo in addition to the combination BRAF-MEK inhibitor therapy. The primary efficacy end point was investigator-assessed PFS per RECIST 1.1 criteria.
At a median follow-up of 19 months, the primary investigator median PFS was 15 months (95% CI, 11.4−18.4) in the atezolizumab arm and 11 months (95% CI, 9.3−12.7) in the placebo arm (HR, 0.78; 95% CI, 0.63−0.97; P = .0249).
An IRC assessment of the triplet therapy found a PFS of 16 months (95% CI, 11.3−18.5), compared with 12 months in the control arm (95% CI, 10.8−14.7) (HR, 0.85; 95% CI, 0.67−1.07).
Objective response rates and complete responses were similar between the treatment groups.
Data are not mature for OS.
Serious adverse events and treatment discontinuations because of toxicity were similar between the arms. Grade 5 adverse events occurred in seven patients in each arm. Two patients with hepatic failure in the atezolizumab group and one patient with pulmonary hemorrhage in the control group were considered treatment related.
The impact of triplet therapy on OS, or when compared with checkpoint inhibitor monotherapy, or to sequential therapy with combination BRAF-MEK inhibitor therapy, preceded or followed by checkpoint inhibition (ongoing trial NCT02224781) is unknown.
Tumor-infiltrating lymphocyte (TIL) therapy
Lifileucel
Lifileucel is an autologous TIL therapy. The FDA approved lifileucel for the treatment of adult patients with unresectable or metastatic melanoma who have previously received a PD-1 blocking antibody and, if BRAF V600 variant–positive, a BRAF inhibitor with or without a MEK inhibitor.
Evidence (lifileucel):
A multicenter, multicohort, open-label trial (C-144-01 [NCT02360579]) evaluated the efficacy and safety of lifileucel in patients with unresectable or metastatic melanoma. The pivotal cohort (cohort 4) included 73 adult patients who had received prior anti–PD-1 therapy and, if BRAF V600 variant–positive, a BRAF inhibitor with or without a MEK inhibitor. Most patients had also received prior ipilimumab. All patients had an ECOG performance status of 0 or 1.
Patients underwent nonmyeloablative lymphodepleting chemotherapy followed by a single infusion of lifileucel and subsequent high-dose IL-2. Tumors were resected and processed to manufacture the autologous TIL product prior to lymphodepletion. Efficacy was assessed according to objective response rate and duration of response.[36][Level of evidence B4]
At a median follow-up of 27.6 months. the objective response rate was 31.5% (95% CI, 21.1%–43.4%), including 4 complete responses and 19 partial responses. The median duration of response was not reached, and 44% of responses lasted 12 months or longer.[36]
In a supporting pooled efficacy set that included 153 patients, the objective response rate was 31.4% (95% CI, 24.1%–39.4%). The median duration of response was not reached, with 54.2% of responses maintained at 12 months.
In a subsequent analysis of the pooled efficacy cohort at a median follow-up of 48.1 months, the median OS was 13.9 months. The 3-year and 4-year OS rates were 28.4% and 21.9%, respectively.[37]
The most common grade 3 or higher adverse events were associated with lymphodepleting chemotherapy and IL-2 administration, including thrombocytopenia, anemia, febrile neutropenia, and hypotension.
Treatment-related mortality was not observed, although high-grade toxicities required inpatient monitoring and supportive care. This is an intense regimen that may only be offered at tertiary centers with experience in its administration.
Lifileucel is given as a one-time treatment and is the first individualized TIL therapy approved for patients with advanced melanoma refractory to PD-1 blockade.
Intralesional therapy
Talimogene laherparepvec (T-VEC)
T-VEC is a genetically modified, herpes simplex virus type 1 (HSV1) oncolytic therapy approved for local intralesional injection into unresectable cutaneous, subcutaneous, and nodal lesions in patients with melanoma that recurs after initial surgery. T-VEC is designed to replicate within tumors, causing lysis, and to produce granulocyte-macrophage colony-stimulating factor (GM-CSF). Release of antigens together with virally derived GM-CSF may promote an antitumor immune response. However, the exact mechanism of action is unknown.
The approval of T-VEC by the FDA was based on data that demonstrated shrinkage of lesions. However, improvement of OS, an effect on visceral metastases, or improvement in quality of life has not been shown.
Evidence (T-VEC):
In a multinational open-label trial (NCT00769704), 436 patients were randomly assigned (2:1) to receive either intralesional T-VEC or subcutaneous GM-CSF for at least 6 months or until there were no more injectable lesions.[38][Level of evidence B3] Eligible patients had stage IIIB, IIIC, or IV melanoma with unresectable, bidimensionally measurable lesions. The primary end point was durable response rate (DRR) (complete response or partial response lasting for >6 months) as assessed by independent review. The study was stratified by site of first recurrence, presence of liver metastases, disease stage, and previous nonadjuvant systemic treatment.
The median patient age was 63 years (range, 22–94), 70% of patients had a baseline ECOG PS score of 0, 30% had stage III disease, and 70% had stage IV disease (27% M1a; 21% M1b; and 22% M1c). Previous therapy for melanoma had been received by 53% of the patients.
The first dose only was administered at 106 plaque-forming units (pfu)/mL to a maximum of 4 mL for all lesions combined. Subsequent doses were administered at 8 pfu/mL up to 4.0 mL for all injected lesions combined with the injected volume based on the size of the lesion. Injection into visceral lesions was not allowed.
In patients treated with T-VEC, 16% (95% CI, 12.0%–20.5%) had a DRR versus 2% (95% CI, 0%–4.5%) in patients who received GM-CSF. Subgroup analysis suggested that the differences in DRRs between T-VEC versus GM-CSF may be greater in earlier-stage disease and treatment-naïve disease, as follows:
In patients with stage IIIB and IIIC disease, the DRR was 33% in the T-VEC group and 0% in the GM-CSF group.
In patients with stage IV M1a disease, the DRR was 16% in the T-VEC group and 2% in the GM-CSF group.
In patients with stage IV M1b disease, the DRR was 3% in the T-VEC group and 4% in the GM-CSF group.
In patients with stage IV M1c disease, the DRR was 7% in the T-VEC group and 3% in the GM-CSF group.
Patients treated with T-VEC or GM-CSF as first-line therapy had a DRR of 24% versus 0%, respectively. However, patients who received treatment as second-line therapy or greater had a DRR of 10% versus 4%, respectively.
The median duration of exposure to T-VEC was 23 weeks (5.3 months), with 26 patients exposed for more than 1 year. The most common adverse events in the T-VEC group were fatigue (50%), chills (49%), pyrexia (43%), nausea (36%), influenza-like illness (30%), and injection site pain. The rate of discontinuation resulting from toxicity to T-VEC was 4%, versus 2% in the GM-CSF group. Of the ten deaths in patients treated with T-VEC, eight deaths were considered the result of PD-1, salmonella infection, and one myocardial infarction; none were considered related to therapy, based on findings of the investigator.
Precautions: T-VEC is a live attenuated HSV and may cause life-threatening, disseminated herpetic infection. It is contraindicated in immunocompromised or pregnant patients. Health care providers and close contacts should avoid direct contact with injected lesions. Biohazard precautions for preparation, administration, and handling are provided in the label.
Detailed prescribing information by treatment cycle and lesion size are provided in the FDA label.
Palliative therapy
Regional lymphadenectomy may be used as palliative care for melanoma that is metastatic to distant, lymph node–bearing areas. Resection may be used as palliative care for isolated metastases to the lung, gastrointestinal tract, bone, or sometimes the brain, with occasional long-term survival.[39–41]
Chemotherapy
Dacarbazine was approved in 1970 based on objective response rates. Phase III trials indicate an objective response rate of 10% to 20%, with rare complete responses observed. An impact on OS has not been demonstrated in randomized trials.[6,21,42–44] When used as a control arm for recent registration trials of ipilimumab and vemurafenib in previously untreated patients with metastatic melanoma, dacarbazine was inferior for OS.
Temozolomide, an oral alkylating agent that hydrolyzes to the same active moiety as dacarbazine, appeared to be similar to dacarbazine (IV administration) in a randomized phase III trial with a primary end point of OS. However, the trial was designed for superiority, and the sample size was inadequate to prove equivalency.[43]
The objective response rate to dacarbazine and the nitrosoureas, carmustine and lomustine, is approximately 10% to 20%.[42,45–47] Responses are usually short-lived, ranging from 3 to 6 months, although long-term remissions can occur in a limited number of patients who attain a complete response.[45,47]
A randomized trial compared IV dacarbazine with temozolomide, an oral agent. OS was 6.4 months for dacarbazine versus 7.7 months for temozolomide (HR, 1.18; 95% CI, 0.92–1.52). While these data suggested similarity between dacarbazine and temozolomide, no benefit in survival has been demonstrated for either dacarbazine or temozolomide. Therefore, evidence of similarity did not result in FDA approval of temozolomide.[43][Level of evidence A1]
An extended schedule and escalated dose of temozolomide was compared with dacarbazine in a multicenter trial by the European Organisation for Research and Treatment of Cancer (EORTC) (EORTC-18032 [NCT00101218]) that randomly assigned 859 patients. No improvement was seen in OS or PFS for the temozolomide group, and this dose and schedule resulted in more toxicity than standard-dose, single-agent dacarbazine.[48][Level of evidence A1]
Two randomized phase III trials in previously untreated patients with metastatic melanoma (resulting in FDA approval for vemurafenib [21] and ipilimumab [6]) included dacarbazine as the standard therapy arm. Both vemurafenib (in BRAF V600–altered melanoma) and ipilimumab showed superior OS compared with dacarbazine in the two separate trials.
Other agents with modest, single-agent activity include vinca alkaloids, platinum compounds, and taxanes.[45,46]
Attempts to develop combination regimens that incorporate chemotherapy (e.g., multiagent chemotherapy,[49,50] combinations of chemotherapy and tamoxifen,[51–53] and combinations of chemotherapy and immunotherapy [9,10,39–41,49,54]) have not demonstrated an improvement in OS.
A published data meta-analysis of 18 randomized trials (15 of which had survival information) that compared chemotherapy with biochemotherapy (i.e., the same chemotherapy plus interferon alone or with IL-2) reported no impact on OS.[55][Level of evidence A1]
Radiation therapy
Although melanoma is a relatively radiation-resistant tumor, palliative radiation therapy may alleviate symptoms. Retrospective studies have shown that symptom relief and some shrinkage of the tumor with radiation therapy may occur in patients with:[56,57]
Multiple brain metastases.
Bone metastases.
Spinal cord compression.
The most effective dose-fractionation schedule for palliation of melanoma metastatic to the bone or spinal cord is unclear, but high-dose-per-fraction schedules are sometimes used to overcome tumor resistance. For more information, visit Cancer Pain.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Robert C, Ribas A, Wolchok JD, et al.: Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 384 (9948): 1109-17, 2014. [PUBMED Abstract]
Robert C, Schachter J, Long GV, et al.: Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 372 (26): 2521-32, 2015. [PUBMED Abstract]
Schachter J, Ribas A, Long GV, et al.: Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 390 (10105): 1853-1862, 2017. [PUBMED Abstract]
Robert C, Long GV, Brady B, et al.: Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372 (4): 320-30, 2015. [PUBMED Abstract]
Zhao X, Suryawanshi S, Hruska M, et al.: Assessment of nivolumab benefit-risk profile of a 240-mg flat dose relative to a 3-mg/kg dosing regimen in patients with advanced tumors. Ann Oncol 28 (8): 2002-2008, 2017. [PUBMED Abstract]
Robert C, Thomas L, Bondarenko I, et al.: Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364 (26): 2517-26, 2011. [PUBMED Abstract]
Hodi FS, O’Day SJ, McDermott DF, et al.: Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363 (8): 711-23, 2010. [PUBMED Abstract]
Yervoy (Ipilimumab): Immune-Mediated Adverse Reaction Management Guide. Princeton, NJ: Bristol-Myers Squibb, 2011. Available online. Last accessed May 1, 2025.
Atkins MB, Lotze MT, Dutcher JP, et al.: High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17 (7): 2105-16, 1999. [PUBMED Abstract]
Atkins MB, Kunkel L, Sznol M, et al.: High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am 6 (Suppl 1): S11-4, 2000. [PUBMED Abstract]
Postow MA, Chesney J, Pavlick AC, et al.: Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372 (21): 2006-17, 2015. [PUBMED Abstract]
Larkin J, Chiarion-Sileni V, Gonzalez R, et al.: Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 373 (1): 23-34, 2015. [PUBMED Abstract]
Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al.: Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 377 (14): 1345-1356, 2017. [PUBMED Abstract]
Wolchok JD, Chiarion-Sileni V, Rutkowski P, et al.: Final, 10-Year Outcomes with Nivolumab plus Ipilimumab in Advanced Melanoma. N Engl J Med 392 (1): 11-22, 2025. [PUBMED Abstract]
Tawbi HA, Forsyth PA, Algazi A, et al.: Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N Engl J Med 379 (8): 722-730, 2018. [PUBMED Abstract]
Tawbi HA, Schadendorf D, Lipson EJ, et al.: Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N Engl J Med 386 (1): 24-34, 2022. [PUBMED Abstract]
Heidorn SJ, Milagre C, Whittaker S, et al.: Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140 (2): 209-21, 2010. [PUBMED Abstract]
Hatzivassiliou G, Song K, Yen I, et al.: RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464 (7287): 431-5, 2010. [PUBMED Abstract]
Poulikakos PI, Zhang C, Bollag G, et al.: RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464 (7287): 427-30, 2010. [PUBMED Abstract]
Su F, Viros A, Milagre C, et al.: RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366 (3): 207-15, 2012. [PUBMED Abstract]
Chapman PB, Hauschild A, Robert C, et al.: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364 (26): 2507-16, 2011. [PUBMED Abstract]
Sosman JA, Kim KB, Schuchter L, et al.: Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 366 (8): 707-14, 2012. [PUBMED Abstract]
Hauschild A, Grob JJ, Demidov LV, et al.: Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380 (9839): 358-65, 2012. [PUBMED Abstract]
Flaherty KT, Robert C, Hersey P, et al.: Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367 (2): 107-14, 2012. [PUBMED Abstract]
Hodi FS, Friedlander P, Corless CL, et al.: Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol 26 (12): 2046-51, 2008. [PUBMED Abstract]
Guo J, Si L, Kong Y, et al.: Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 29 (21): 2904-9, 2011. [PUBMED Abstract]
Carvajal RD, Antonescu CR, Wolchok JD, et al.: KIT as a therapeutic target in metastatic melanoma. JAMA 305 (22): 2327-34, 2011. [PUBMED Abstract]
Long GV, Stroyakovskiy D, Gogas H, et al.: Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371 (20): 1877-88, 2014. [PUBMED Abstract]
Long GV, Stroyakovskiy D, Gogas H, et al.: Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386 (9992): 444-51, 2015. [PUBMED Abstract]
Robert C, Karaszewska B, Schachter J, et al.: Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372 (1): 30-9, 2015. [PUBMED Abstract]
Robert C, Grob JJ, Stroyakovskiy D, et al.: Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med 381 (7): 626-636, 2019. [PUBMED Abstract]
Larkin J, Ascierto PA, Dréno B, et al.: Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371 (20): 1867-76, 2014. [PUBMED Abstract]
Dummer R, Ascierto PA, Gogas HJ, et al.: Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 19 (5): 603-615, 2018. [PUBMED Abstract]
Dummer R, Ascierto PA, Gogas HJ, et al.: Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 19 (10): 1315-1327, 2018. [PUBMED Abstract]
Gutzmer R, Stroyakovskiy D, Gogas H, et al.: Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 395 (10240): 1835-1844, 2020. [PUBMED Abstract]
Chesney J, Lewis KD, Kluger H, et al.: Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: pooled analysis of consecutive cohorts of the C-144-01 study. J Immunother Cancer 10 (12): , 2022. [PUBMED Abstract]
Medina T, Chesney JA, Whitman E, et al.: Long-term efficacy and safety of lifileucel tumor-infiltrating lymphocyte (TIL) cell therapy in patients with advanced melanoma: a 4-year analysis of the C-144–01 study. [Abstract] J Immunother Cancer 11 (Suppl 1): A-776, 873, 2023.
Andtbacka RH, Kaufman HL, Collichio F, et al.: Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 33 (25): 2780-8, 2015. [PUBMED Abstract]
Leo F, Cagini L, Rocmans P, et al.: Lung metastases from melanoma: when is surgical treatment warranted? Br J Cancer 83 (5): 569-72, 2000. [PUBMED Abstract]
Ollila DW, Hsueh EC, Stern SL, et al.: Metastasectomy for recurrent stage IV melanoma. J Surg Oncol 71 (4): 209-13, 1999. [PUBMED Abstract]
Gutman H, Hess KR, Kokotsakis JA, et al.: Surgery for abdominal metastases of cutaneous melanoma. World J Surg 25 (6): 750-8, 2001. [PUBMED Abstract]
Chapman PB, Einhorn LH, Meyers ML, et al.: Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol 17 (9): 2745-51, 1999. [PUBMED Abstract]
Middleton MR, Grob JJ, Aaronson N, et al.: Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 18 (1): 158-66, 2000. [PUBMED Abstract]
Avril MF, Aamdal S, Grob JJ, et al.: Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: a phase III study. J Clin Oncol 22 (6): 1118-25, 2004. [PUBMED Abstract]
Anderson CM, Buzaid AC, Legha SS: Systemic treatments for advanced cutaneous melanoma. Oncology (Huntingt) 9 (11): 1149-58; discussion 1163-4, 1167-8, 1995. [PUBMED Abstract]
Wagner JD, Gordon MS, Chuang TY, et al.: Current therapy of cutaneous melanoma. Plast Reconstr Surg 105 (5): 1774-99; quiz 1800-1, 2000. [PUBMED Abstract]
Mays SR, Nelson BR: Current therapy of cutaneous melanoma. Cutis 63 (5): 293-8, 1999. [PUBMED Abstract]
Patel PM, Suciu S, Mortier L, et al.: Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur J Cancer 47 (10): 1476-83, 2011. [PUBMED Abstract]
Kirkwood JM, Strawderman MH, Ernstoff MS, et al.: Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14 (1): 7-17, 1996. [PUBMED Abstract]
Kirkwood JM, Ibrahim JG, Sondak VK, et al.: High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol 18 (12): 2444-58, 2000. [PUBMED Abstract]
Kirkwood JM, Ibrahim J, Lawson DH, et al.: High-dose interferon alfa-2b does not diminish antibody response to GM2 vaccination in patients with resected melanoma: results of the Multicenter Eastern Cooperative Oncology Group Phase II Trial E2696. J Clin Oncol 19 (5): 1430-6, 2001. [PUBMED Abstract]
Hancock BW, Wheatley K, Harris S, et al.: Adjuvant interferon in high-risk melanoma: the AIM HIGH Study–United Kingdom Coordinating Committee on Cancer Research randomized study of adjuvant low-dose extended-duration interferon Alfa-2a in high-risk resected malignant melanoma. J Clin Oncol 22 (1): 53-61, 2004. [PUBMED Abstract]
Koops HS, Vaglini M, Suciu S, et al.: Prophylactic isolated limb perfusion for localized, high-risk limb melanoma: results of a multicenter randomized phase III trial. European Organization for Research and Treatment of Cancer Malignant Melanoma Cooperative Group Protocol 18832, the World Health Organization Melanoma Program Trial 15, and the North American Perfusion Group Southwest Oncology Group-8593. J Clin Oncol 16 (9): 2906-12, 1998. [PUBMED Abstract]
Lee ML, Tomsu K, Von Eschen KB: Duration of survival for disseminated malignant melanoma: results of a meta-analysis. Melanoma Res 10 (1): 81-92, 2000. [PUBMED Abstract]
Ives NJ, Stowe RL, Lorigan P, et al.: Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2,621 patients. J Clin Oncol 25 (34): 5426-34, 2007. [PUBMED Abstract]
Rate WR, Solin LJ, Turrisi AT: Palliative radiotherapy for metastatic malignant melanoma: brain metastases, bone metastases, and spinal cord compression. Int J Radiat Oncol Biol Phys 15 (4): 859-64, 1988. [PUBMED Abstract]
Herbert SH, Solin LJ, Rate WR, et al.: The effect of palliative radiation therapy on epidural compression due to metastatic malignant melanoma. Cancer 67 (10): 2472-6, 1991. [PUBMED Abstract]
Latest Updates to This Summary (05/02/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Revised text about the results of a trial that randomly assigned patients with completely resected stage IIB or stage IIC melanoma to receive either pembrolizumab or placebo (cited Luke et al. as reference 19).
Revised text about the final analysis results of a multinational double-blind trial that randomly assigned patients with stage IIIA, IIIB, or IIIC melanoma with BRAF V600E or V600K pathogenic variants who underwent completion lymphadenectomy to receive either dabrafenib plus trametinib or two matched placebo tablets (cited Long et al. as reference 32).
Revised the list of treatment options for unresectable stage III, stage IV, and recurrent melanoma to include tumor-infiltrating lymphocyte therapy.
Revised text about the final analysis results of an international double-blind trial that randomly assigned 945 previously untreated patients with unresectable stage III or IV melanoma to receive nivolumab plus placebo, nivolumab plus ipilimumab followed by nivolumab, or ipilimumab plus placebo (cited Wolchok et al. as reference 14).
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of melanoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Melanoma Treatment is:
Shaheer A. Khan, DO (Columbia University Irving Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Melanoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/skin/hp/melanoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389469]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Merkel cell carcinoma is a very rare type of cancer that forms in the skin.
Sun exposure and having a weak immune system affects the risk of developing Merkel cell carcinoma.
Merkel cell carcinoma usually appears as a single painless lump on sun-exposed skin.
Tests that examine the skin are used to diagnose Merkel cell carcinoma.
After Merkel cell carcinoma has been diagnosed, tests are done to find out if cancer cells have spread to other parts of the body.
Some people decide to get a second opinion.
Certain factors affect prognosis (chance of recovery) and treatment options.
Merkel cell carcinoma is a very rare type of cancer that forms in the skin.
Merkel cells are found in the top layer of the skin. These cells are very close to the nerve endings that receive the sensation of touch. Merkel cell carcinoma, also called neuroendocrine carcinoma of the skin or trabecular cancer, is a very rare type of skin cancer that forms when Merkel cells grow out of control. Merkel cell carcinoma starts most often in areas of skin exposed to the sun, especially the head and neck, as well as the arms, legs, and trunk.
EnlargeAnatomy of the skin showing the epidermis, dermis, and subcutaneous tissue. Merkel cells are in the layer of basal cells at the deepest part of the epidermis and are connected to nerves.
Merkel cell carcinoma tends to grow quickly and to metastasize (spread) at an early stage. It usually spreads first to nearby lymph nodes and then may spread to lymph nodes or skin in distant parts of the body, lungs, brain, bones, or other organs.
Merkel cell carcinoma is the second most common cause of skin cancer death after melanoma.
Sun exposure and having a weak immune system affects the risk of developing Merkel cell carcinoma.
Anything that increases a person’s chance of getting a disease is called a risk factor. Not every person with one or more of these risk factors will develop Merkel cell carcinoma, and it can develop in people who don’t have any known risk factors. Talk with your doctor if you think you may be at risk.
Merkel cell carcinoma usually appears as a single painless lump on sun-exposed skin.
This and other changes in the skin may be caused by Merkel cell carcinoma or by other conditions. Check with your doctor if you see changes in your skin.
Merkel cell carcinoma usually appears on sun-exposed skin as a single lump that is:
fast-growing
painless
firm and dome-shaped or raised
red or violet in color
Tests that examine the skin are used to diagnose Merkel cell carcinoma.
Full-body skin exam is an exam of the skin. A doctor or nurse checks the skin for bumps or spots that look abnormal in color, size, shape, or texture. The size, shape, and texture of the lymph nodes will also be checked.
Skin biopsy is the removal of skin cells or tissues so they can be viewed under a microscope by a pathologist to check for signs of cancer.
After Merkel cell carcinoma has been diagnosed, tests are done to find out if cancer cells have spread to other parts of the body.
The process used to find out if cancer has spread to other parts of the body is called staging. The information gathered from the staging process determines the stage of the disease. It is important to know the stage in order to plan treatment.
The following tests and procedures may be used in the staging process:
CT scan (CAT scan) uses a computer linked to an x-ray machine to make a series of detailed pictures of areas inside the body, such as the chest, abdomen, head, and neck. The pictures are taken from different angles and are used to create 3-D views of tissues and organs. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography. Learn more about Computed Tomography (CT) Scans and Cancer.
PET scan (positron emission tomography scan) uses a small amount of radioactive sugar (also called radioactive glucose) that is injected into a vein. The PET scanner rotates around the body and makes pictures of where sugar is being used by the body. Cancer cells show up brighter in the pictures because they are more active and take up more sugar than normal cells do.
Lymph node biopsy is the removal of all or part of a lymph node. A pathologist views the lymph node tissue under a microscope to check for cancer cells. This procedure is also called lymph node sampling. There are several types of lymph node biopsy used to stage Merkel cell carcinoma.
Sentinel lymph node biopsy removes the sentinel lymph node during surgery. The sentinel lymph node is the first lymph node in a group of lymph nodes to receive lymphatic drainage from the primary tumor. It is therefore the first lymph node the cancer is likely to spread to from the primary tumor. To identify the sentinel lymph node, a radioactive substance, blue dye, or both is injected near the tumor. The substance or dye flows through the lymphducts to the lymph nodes. The first lymph node to receive the substance or dye is removed. A pathologist views the tissue under a microscope to look for cancer cells. If cancer cells are found, more lymph nodes will be removed through a separate incision (cut). This is called a lymph node dissection. Sometimes, a sentinel lymph node is found in more than one group of nodes. EnlargeSentinel lymph node biopsy of the skin. A radioactive substance and/or blue dye is injected near the tumor (first panel). The injected material is detected visually and/or with a probe that detects radioactivity (middle panel). The sentinel nodes (the first lymph nodes to take up the material) are removed and checked for cancer cells (last panel).
Lymph node dissection is a surgical procedure in which the lymph nodes are removed and a sample of tissue is checked under a microscope for signs of cancer. For a regional lymph node dissection, some of the lymph nodes in the tumor area are removed. For a radical lymph node dissection, most or all of the lymph nodes in the tumor area are removed. This procedure is also called lymphadenectomy.
Core needle biopsy is a procedure to remove a sample of tissue using a wide needle. A pathologist views the tissue under a microscope to look for cancer cells.
Fine-needle aspiration biopsy is a procedure to remove a sample of tissue using a thin needle. A pathologist views the tissue under a microscope to look for cancer cells.
Immunohistochemistry uses antibodies to check for certain antigens (markers) in a sample of a patient’s cells or tissue. The antibodies are usually linked to an enzyme or a fluorescent dye. After the antibodies bind to a specific antigen in the tissue sample, the enzyme or dye is activated, and the antigen can then be seen under a microscope. This type of test is used to help diagnose cancer and help tell one type of cancer from another type.
Some people decide to get a second opinion.
You may want to get a second opinion to confirm your Merkel cell carcinoma diagnosis and treatment plan. If you seek a second opinion, you will need to get medical test results and reports from the first doctor to share with the second doctor. The second doctor will review the pathology report, slides, and scans. They may agree with the first doctor, suggest changes or another treatment approach, or provide more information about your cancer.
Certain factors affect prognosis (chance of recovery) and treatment options.
The prognosis and treatment options depend on:
the stage of the cancer (the size of the tumor and whether it has spread to the lymph nodes or other parts of the body)
where the cancer is in the body
whether the cancer has just been diagnosed or has recurred (come back)
the person’s age and general health
Prognosis also depends on how deeply the tumor has grown into the skin.
Stages of Merkel Cell Carcinoma
Key Points
The following stages are used for Merkel cell carcinoma:
Stage 0 (carcinoma in situ)
Stage I (also called Stage 1) Merkel cell carcinoma
Stage II (also called Stage 2) Merkel cell carcinoma
Stage III (also called Stage 3) Merkel cell carcinoma
Stage IV (also called Stage 4) Merkel cell carcinoma
It is common for Merkel cell carcinoma to recur (come back) after it has been treated.
Cancer stage describes the extent of cancer in the body, such as the size of the tumor, whether it has spread, and how far it has spread from where it first formed. It is important to know the stage of Merkel cell cancer to plan the best treatment.
There are several staging systems for cancer that describe the extent of the cancer. Merkel cell carcinoma staging usually uses the TNM staging system. The cancer may be described by this staging system in your pathology report. Based on the TNM results, a stage (I, II, III, or IV, also written as 1, 2, 3, or 4) is assigned to your cancer. When talking to you about your diagnosis, your doctor may describe the cancer as one of these stages.
The following stages are used for Merkel cell carcinoma:
EnlargeTumor sizes are often measured in centimeters (cm) or inches. Common food items that can be used to show tumor size in cm include: a pea (1 cm), a peanut (2 cm), a grape (3 cm), a walnut (4 cm), a lime (5 cm or 2 inches), an egg (6 cm), a peach (7 cm), and a grapefruit (10 cm or 4 inches).
Stage 0 (carcinoma in situ)
In stage 0, abnormalMerkel cells are found in the top layer of skin. These abnormal cells may become cancer and spread into nearby normal tissue.
Stage I (also called Stage 1) Merkel cell carcinoma
In stage I, the tumor is 2 centimeters or smaller.
Stage II (also called Stage 2) Merkel cell carcinoma
Stage II Merkel cell carcinoma is divided into stages IIA and IIB.
In stage IIA, the tumor is larger than 2 centimeters.
Stage III (also called Stage 3) Merkel cell carcinoma
Stage III Merkel cell carcinoma is divided into stages IIIA and IIIB.
In stage IIIA, either of the following is found:
the tumor may be any size and may have spread to nearby connective tissue, muscle, cartilage, or bone. A lymph node cannot be felt during a physical exam but cancer is found in the lymph node by sentinel lymph node biopsy or after the lymph node is removed and checked under a microscope for signs of cancer; or
a swollen lymph node is felt during a physical exam and/or seen on an imaging test. When the lymph node is removed and checked under a microscope for signs of cancer, cancer is found in the lymph node. The place where the cancer began is not known.
In stage IIIB, the tumor may be any size and:
may have spread to nearby connective tissue, muscle, cartilage, or bone. A swollen lymph node is felt during a physical exam and/or seen on an imaging test. When the lymph node is removed and checked under a microscope for signs of cancer, cancer is found in the lymph node; or
cancer is in a lymph vessel between the primary tumor and lymph nodes that are near or far away. Cancer may have spread to lymph nodes.
Stage IV (also called Stage 4) Merkel cell carcinoma
In stage IV, the tumor has spread to skin that is not close to the primary tumor or to other parts of the body, such as the liver, lung, bone, or brain.
Stage IV Merkel cell carcinoma is also called metastatic Merkel cell carcinoma. Metastatic cancer happens when cancer cells travel through the lymphatic system or blood and form tumors in other parts of the body. The metastatic tumor is the same type of cancer as the primary tumor. For example, if Merkel cell carcinoma spreads to the liver, the cancer cells in the liver are actually Merkel cell carcinoma cells. The disease is called metastatic Merkel cell carcinoma, not liver cancer. Learn more in Metastatic Cancer: When Cancer Spreads.
It is common for Merkel cell carcinoma to recur (come back) after it has been treated.
Recurrent Merkel cell carcinoma is cancer that has come back after it has been treated. If Merkel cell carcinoma comes back, it may come back in the skin, lymph nodes, or other parts of the body. Tests will be done to help determine where the cancer has returned. The type of treatment for recurrent Merkel cell carcinoma will depend on where it has come back.
There are different types of treatment for people with Merkel cell carcinoma.
The following types of treatment are used:
Surgery
Radiation therapy
Chemotherapy
Immunotherapy
New types of treatment are being tested in clinical trials.
Treatment for Merkel cell carcinoma may cause side effects.
Follow-up care may be needed.
There are different types of treatment for people with Merkel cell carcinoma.
Different types of treatments are available for Merkel cell carcinoma. You and your cancer care team will work together to decide your treatment plan, which may include more than one type of treatment. Many factors will be considered, such as the stage of the cancer, your overall health, and your preferences. Your plan will include information about your cancer, the goals of treatment, your treatment options and the possible side effects, and the expected length of treatment.
Talking with your cancer care team before treatment begins about what to expect will be helpful. You’ll want to learn what you need to do before treatment begins, how you’ll feel while going through it, and what kind of help you will need. To learn more, visit Questions to Ask Your Doctor About Treatment.
The following types of treatment are used:
Surgery
One or more of the following surgical procedures may be used to treat Merkel cell carcinoma:
Lymph node dissection: A surgical procedure in which the lymph nodes are removed and a sample of tissue is checked under a microscope for signs of cancer. For a regional lymph node dissection, some of the lymph nodes in the tumor area are removed; for a radical lymph node dissection, most or all of the lymph nodes in the tumor area are removed. This procedure is also called lymphadenectomy.
After the doctor removes all the cancer that can be seen at the time of the surgery, some patients may be given chemotherapy or radiation therapy after surgery to kill any cancer cells that are left. Treatment given after the surgery, to lower the risk that the cancer will come back, is called adjuvant therapy.
Radiation therapy
Radiation therapy uses high-energy x-rays or other types of radiation to kill cancer cells or keep them from growing. External radiation therapy uses a machine outside the body to send radiation toward the area of the body with cancer. It is used to treat Merkel cell carcinoma and may also be used as palliative therapy to relieve symptoms and improve quality of life.
Chemotherapy (also called chemo) uses drugs to stop the growth of cancer cells, either by killing the cells or by stopping the cells from dividing. When chemotherapy is taken by mouth or injected into a vein or muscle, the drugs enter the bloodstream and can reach cancer cells throughout the body (systemic chemotherapy).
New types of treatment are being tested in clinical trials.
For some people, joining a clinical trial may be an option. There are different types of clinical trials for people with cancer. For example, a treatment trial tests new treatments or new ways of using current treatments. Supportive care and palliative care trials look at ways to improve quality of life, especially for those who have side effects from cancer and its treatment.
You can use the clinical trial search to find NCI-supported cancer clinical trials accepting participants. The search allows you to filter trials based on the type of cancer, your age, and where the trials are being done. Clinical trials supported by other organizations can be found on the ClinicalTrials.gov website.
As you go through treatment, you will have follow-up tests or check-ups. Some tests that were done to diagnose or stage the cancer may be repeated to see how well the treatment is working. Decisions about whether to continue, change, or stop treatment may be based on the results of these tests.
Some of the tests will continue to be done from time to time after treatment has ended. The results of these tests can show if your condition has changed or if the cancer has recurred (come back).
Treatment of Stage I and Stage II Merkel Cell Carcinoma
Treatment of stage I and stage II Merkel cell carcinoma may include:
Use our clinical trial search to find NCI-supported cancer clinical trials that are accepting patients. You can search for trials based on the type of cancer, the age of the patient, and where the trials are being done. General information about clinical trials is also available.
Treatment of Stage III Merkel Cell Carcinoma
Treatment of stage III Merkel cell carcinoma may include:
Use our clinical trial search to find NCI-supported cancer clinical trials that are accepting patients. You can search for trials based on the type of cancer, the age of the patient, and where the trials are being done. General information about clinical trials is also available.
Treatment of Stage IV Merkel Cell Carcinoma
Treatment of stage IV Merkel cell carcinoma may include:
Use our clinical trial search to find NCI-supported cancer clinical trials that are accepting patients. You can search for trials based on the type of cancer, the age of the patient, and where the trials are being done. General information about clinical trials is also available.
Treatment of Recurrent Merkel Cell Carcinoma
Treatment of recurrent Merkel cell carcinoma may include:
Use our clinical trial search to find NCI-supported cancer clinical trials that are accepting patients. You can search for trials based on the type of cancer, the age of the patient, and where the trials are being done. General information about clinical trials is also available.
Physician Data Query (PDQ) is the National Cancer Institute’s (NCI’s) comprehensive cancer information database. The PDQ database contains summaries of the latest published information on cancer prevention, detection, genetics, treatment, supportive care, and complementary and alternative medicine. Most summaries come in two versions. The health professional versions have detailed information written in technical language. The patient versions are written in easy-to-understand, nontechnical language. Both versions have cancer information that is accurate and up to date and most versions are also available in Spanish.
PDQ is a service of the NCI. The NCI is part of the National Institutes of Health (NIH). NIH is the federal government’s center of biomedical research. The PDQ summaries are based on an independent review of the medical literature. They are not policy statements of the NCI or the NIH.
Purpose of This Summary
This PDQ cancer information summary has current information about the treatment of merkel cell carcinoma. It is meant to inform and help patients, families, and caregivers. It does not give formal guidelines or recommendations for making decisions about health care.
Reviewers and Updates
Editorial Boards write the PDQ cancer information summaries and keep them up to date. These Boards are made up of experts in cancer treatment and other specialties related to cancer. The summaries are reviewed regularly and changes are made when there is new information. The date on each summary (“Updated”) is the date of the most recent change.
The information in this patient summary was taken from the health professional version, which is reviewed regularly and updated as needed, by the PDQ Adult Treatment Editorial Board.
Clinical Trial Information
A clinical trial is a study to answer a scientific question, such as whether one treatment is better than another. Trials are based on past studies and what has been learned in the laboratory. Each trial answers certain scientific questions in order to find new and better ways to help cancer patients. During treatment clinical trials, information is collected about the effects of a new treatment and how well it works. If a clinical trial shows that a new treatment is better than one currently being used, the new treatment may become “standard.” Patients may want to think about taking part in a clinical trial. Some clinical trials are open only to patients who have not started treatment.
Clinical trials can be found online at NCI’s website. For more information, call the Cancer Information Service (CIS), NCI’s contact center, at 1-800-4-CANCER (1-800-422-6237).
Permission to Use This Summary
PDQ is a registered trademark. The content of PDQ documents can be used freely as text. It cannot be identified as an NCI PDQ cancer information summary unless the whole summary is shown and it is updated regularly. However, a user would be allowed to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks in the following way: [include excerpt from the summary].”
The best way to cite this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Merkel Cell Carcinoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/skin/patient/merkel-cell-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389202]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use in the PDQ summaries only. If you want to use an image from a PDQ summary and you are not using the whole summary, you must get permission from the owner. It cannot be given by the National Cancer Institute. Information about using the images in this summary, along with many other images related to cancer can be found in Visuals Online. Visuals Online is a collection of more than 3,000 scientific images.
Disclaimer
The information in these summaries should not be used to make decisions about insurance reimbursement. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s E-mail Us.
Merkel cell carcinoma (MCC) was originally described by Toker in 1972 as trabecular carcinoma of the skin.[1] Other names for MCC include Toker tumor, primary small cell carcinoma of the skin, primary cutaneous neuroendocrine tumor, and malignant trichodiscoma.[2]
MCC is an aggressive neuroendocrine carcinoma arising in the dermoepidermal junction (see Figure 1), and it is the second most common cause of skin cancer death after melanoma.[3,4] Although the exact origin and function of the Merkel cell remains under investigation, it is thought to have features of both epithelial and neuroendocrine origin and arise in cells with touch-sensitivity function (mechanoreceptors).[5–11]
Therapeutic options have been historically limited for patients with advanced disease. However, new immunotherapeutic approaches are associated with durable responses.[12]
MCC incidence increases progressively with age. There are few cases in patients younger than 50 years, and the median age at diagnosis is about 65 years (see Figure 2).[13] Incidence is considerably greater in White individuals than in Black individuals and slightly greater in men than in women.[13–17]
EnlargeFigure 2. Frequency of MCC by age and sex of men (square) and women (circle). Reprinted from Journal of the American Academy of Dermatology, 49 (5), Agelli M and Clegg L, Epidemiology of primary Merkel cell carcinoma in the United States, pp. 832–41, Copyright (2003), with permission from Elsevier.
MCC occurs most frequently in sun-exposed areas of skin, particularly the head and neck, followed by the extremities, and then the trunk.[5,16,18] Incidence has been reported to be greater in geographic regions with higher levels of ultraviolet B sunlight.[16]
As of 2013, MCC had an annual incidence of 0.7 cases per 100,000 people in the United States.[19] The incidence has been increasing over the past several decades, almost doubling in the United States between 2000 and 2013. This rise is potentially related to more accurate diagnostic pathology tools, improved clinical awareness of MCC, an aging population, increased sun exposure in susceptible populations, and improved registry tools. The incidence is also higher in immunosuppressed populations (HIV, hematologic malignancies, immunosuppressive medications, etc.).[20] Approximately 25,000 cases of MCC have been recorded in the United States since 2000, including more than 2,200 incident cases reported in 2014 to the National Program of Cancer Registries/SEER combined registries, which captures more than 98% of the U.S. population and the ten most common sites of MCC (see Table 1).[17]
Table 1. The Ten Most Common Sites for Merkel Cell Carcinoma (SEER 1973–2006)a
Anatomical Site
Cases (%)
NOS = not otherwise specified; SEER = Surveillance, Epidemiology, and End Results Program.
In various cases series, up to 97% of MCCs arise in the skin. MCC primaries in other sites were very rare, as were MCCs from unknown primary sites.[17]
SEER registry data have shown excess risk of MCC as a first or second cancer in patients with several primary cancers.[21] National cancer registries from three Scandinavian countries have identified a variety of second cancers diagnosed after MCC.[22]
Pathogenesis
Increased incidence of MCC has also been seen in people treated heavily with methoxsalen (psoralen) and ultraviolet A (PUVA) for psoriasis (3 of 1,380 patients, 0.2%). This has also been seen in individuals with chronic immune suppression, especially from chronic lymphocytic leukemia, HIV, and previous solid organ transplant.[16,23]
In 2008, a novel polyomavirus (Merkel cell polyoma virus [MCPyV]) was first reported in MCC tumor specimens,[24] a finding subsequently confirmed in other laboratories.[25–27] High levels of viral DNA and clonal integration of the virus in MCC tumors have also been reported, [28] along with expression of certain viral antigens in MCC cells and the presence of antiviral antibodies. Not all cases of MCC appear to be associated with MCPyV infection.[29]
MCPyV has been detected at very low levels in normal skin distant from the MCC primary tumor, in a significant percentage of patients with non-MCC cutaneous disorders, in normal-appearing skin in healthy individuals, and in nonmelanoma skin cancers in immune-suppressed individuals.[10,30–32] Various methods have been used to identify and quantify the presence of MCPyV in MCC tumor specimens, other non-MCC tumors, blood, urine, and other tissues.[33,34]
The significance of the new MCPyV findings remains uncertain. The prognostic significance of viral load, antibody titer levels, and the role of underlying immunosuppression in hosts (from disease and medications) are under investigation.
Prevalence of MCPyV appears to differ between MCC patients in the United States and Europe versus Australia. There may be two independent pathways for the development of MCC: one driven by the presence of MCPyV, and the other driven primarily by sun damage, especially as noted in patient series from Australia.[25,29,35]
Although no unique marker for MCC has been identified, a variety of molecular and cytogenetic markers of MCC have been reported.[7,10,36]
Clinical Presentation
MCC usually presents as a painless, indurated, solitary dermal nodule with a slightly erythematous to deeply violaceous color, and rarely, an ulcer. MCC can infiltrate locally via dermal lymphatics, resulting in multiple satellite lesions. Because of its nonspecific clinical appearance, MCC is rarely suspected before a biopsy is performed.[5] Photographs of MCC skin lesions illustrate its clinical variability.[37]
A mnemonic [18] summarizing typical clinical characteristics of MCC has been proposed:
AEIOU
A = Asymptomatic.
E = Expanding rapidly.
I = Immune suppressed.
O = Older than 50 years.
U = UV-exposed skin.
Not all patients have every element in this mnemonic. However, in this study, 89% of patients met three or more criteria, 52% met four or more criteria, and 7% met all five criteria.[18]
Initial Clinical Evaluation
Because local-regional spread is common, patients with newly diagnosed MCC require a careful clinical examination for satellite lesions and regional nodal involvement.
Tailoring an imaging work-up to the clinical presentation and any relevant signs and symptoms should be considered. There has been no systematic study of the optimal imaging work-up for newly diagnosed patients, and it is not clear if all newly diagnosed patients, especially those with the smallest primary tumors, benefit from a detailed imaging work-up.
If an imaging work-up is performed, it may include a computed tomography (CT) scan of the chest and abdomen to rule out primary small cell lung cancer as well as distant and regional metastases. Imaging studies designed to evaluate suspicious signs and symptoms may also be recommended. In one series, CT scans had an 80% false-negative rate for regional metastases.[38] Head and neck presentations may require additional imaging. Magnetic resonance imaging has been used to evaluate MCC but has not been studied systematically.[39] Fluorine F 18-fludeoxyglucose positron emission tomography results have been reported in selected cases.[40,41] Baseline routine blood work has been recommended but has not been studied systematically. There are no known circulating tumor markers specific to MCC.
Initial Staging Results
The results of initial clinical staging of MCC vary widely in the literature, based on retrospective case series reported over decades. For invasive cancers, 48.6% were localized, 31.1% were regional, and 8.2% were distant.[17]
MCC that presents in regional nodes without an identifiable primary lesion is found in a minority of patients, with the percent of these cases varying among the reported series. Tumors without an identifiable primary lesion have been attributed to either spontaneous regression of the primary or metastatic neuroendocrine carcinoma from a clinically occult site.[8,17,18,42,43]
Clinical Progression
In a review of patients from 18 case series, 279 of 926 patients (30.1%) developed local recurrence during follow-up, excluding those presenting with distant metastatic disease. These events have been typically attributed to inadequate surgical margins and/or a lack of adjuvant radiation therapy. In addition, 545 of 982 patients (55.5%) had lymph node metastases at diagnosis or during follow-up.[8]
In the same review of 18 case series, the most common sites of distant metastases were distant lymph nodes (60.1%), distant skin (30.3%), lung (23.4%), central nervous system (18.4%), and bone (15.2%).[8] Many other sites of disease have also been reported, and the distribution of metastatic sites varies among case series.
In one series of 237 patients presenting with local or regional disease, the median time-to-recurrence was 9 months (range, 2–70 months). Ninety-one percent of recurrences occurred within 2 years of diagnosis.[44]
Potential Prognostic Factors
The extent of disease at presentation may provide the most useful estimate of prognosis.[7]
Diagnostic procedures, such as sentinel lymph node biopsy, may help distinguish between local and regional disease at presentation. One-third of patients who lack clinically palpable or radiologically visible nodes will have microscopically evident regional disease.[38] Nodal positivity may be substantially lower among patients with small tumors (e.g., ≤1.0 cm).[45]
Many retrospective studies have evaluated the relationship of a wide variety of biological and histological factors to survival and local-regional control.[7,8,17,38,44,46–57][Level of evidence C2] Many of these reports are confounded by small numbers, potential selection bias, referral bias, short follow-up, no uniform clinical protocol for both staging and treatment, and are underpowered to detect modest differences.
A large, single-institution, retrospective study of 156 patients with MCC, with a median follow-up of 51 months (range, 2–224 months), evaluated histological factors potentially associated with prognosis.[55][Level of evidence C1] Although this report was subject to potential selection and referral bias, both univariate and multivariate analyses demonstrated a relationship between improved cause-specific survival and circumscribed growth pattern versus infiltrative pattern, shallow-tumor depth versus deep-tumor depth, and absence of lymphovascular invasion versus presence of lymphovascular invasion. Adoption of these findings into a global prognostic algorithm awaits independent confirmation by adequately powered studies.
A 2009 study investigated whether the presence of newly identified MCPyV in MCC tumor specimens influenced clinical outcome among 114 Finnish patients with MCC. In this small study, patients whose tumors were MCPyV positive appeared to have better survival than patients whose tumors were MCPyV negative.[58][Level of evidence C2] Standardization of procedures to identify and quantify MCPyV and relevant antibodies is needed to improve understanding of both prognostic and epidemiological questions.[10]
Prognosis
The most significant prognostic parameters for MCC include tumor size and the presence of locoregional or distant metastases. These factors form the basis of the American Joint Committee on Cancer staging system for MCC.[59,60] Although an increasing primary tumor size correlates with an increased risk of metastatic disease, MCC tumors of any size have significant risk of occult metastasis, supporting the use of sentinel lymph node biopsy for all cases.[61] Additional features of the primary tumor, such as lymphovascular invasion and tumor growth pattern, may also have prognostic significance. Clinically detectable nodal disease is associated with worse outcome than microscopic metastases.[55,59] Other findings associated with worse prognosis include sheet-like involvement in lymph node metastases and an increasing number of metastatic lymph nodes.[60,62]
The bulk of MCC literature is from small case series, which are subject to many confounding factors. For this reason, the relapse and survival rates reported by stage vary widely in the literature. In general, lower-stage disease is associated with better overall survival.[63] For more information, see the Potential Prognostic Factors section.
Outcomes from patients presenting with small volume local disease and pathologically confirmed cancer-negative lymph nodes report a cause-specific 5-year survival rate exceeding 90% in one report.[44,55][Level of evidence C2]
A tabular summary of treatment results of MCC from 12 series illustrates the difficulty in comparing outcome data among series.[7]
Using the SEER Program registry MCC staging system adopted in 1973, MCC survival data (1973–2006) by stage is summarized in Figure 3.[17]
Toker C: Trabecular carcinoma of the skin. Arch Dermatol 105 (1): 107-10, 1972. [PUBMED Abstract]
Schwartz RA, Lambert WC: The Merkel cell carcinoma: a 50-year retrospect. J Surg Oncol 89 (1): 5, 2005. [PUBMED Abstract]
Agelli M, Clegg LX, Becker JC, et al.: The etiology and epidemiology of merkel cell carcinoma. Curr Probl Cancer 34 (1): 14-37, 2010 Jan-Feb. [PUBMED Abstract]
Harms PW: Update on Merkel Cell Carcinoma. Clin Lab Med 37 (3): 485-501, 2017. [PUBMED Abstract]
Nghiem P, James N: Merkel cell carcinoma. In: Wolff K, Goldsmith LA, Katz SI, et al., eds.: Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill , 2008, pp 1087-94.
Eng TY, Boersma MG, Fuller CD, et al.: A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol 30 (6): 624-36, 2007. [PUBMED Abstract]
Medina-Franco H, Urist MM, Fiveash J, et al.: Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol 8 (3): 204-8, 2001. [PUBMED Abstract]
Busse PM, Clark JR, Muse VV, et al.: Case records of the Massachusetts General Hospital. Case 19-2008. A 63-year-old HIV-positive man with cutaneous Merkel-cell carcinoma. N Engl J Med 358 (25): 2717-23, 2008. [PUBMED Abstract]
Rockville Merkel Cell Carcinoma Group: Merkel cell carcinoma: recent progress and current priorities on etiology, pathogenesis, and clinical management. J Clin Oncol 27 (24): 4021-6, 2009. [PUBMED Abstract]
Calder KB, Smoller BR: New insights into merkel cell carcinoma. Adv Anat Pathol 17 (3): 155-61, 2010. [PUBMED Abstract]
Young JL, Ward KC, Ries LAG: Cancer of rare sites. In: Ries LAG, Young JL, Keel GE, et al., eds.: SEER Survival Monograph: Cancer Survival Among Adults: U. S. SEER Program, 1988-2001, Patient and Tumor Characteristics. National Cancer Institute, 2007. NIH Pub. No. 07-6215, pp 251-61.
Miller RW, Rabkin CS: Merkel cell carcinoma and melanoma: etiological similarities and differences. Cancer Epidemiol Biomarkers Prev 8 (2): 153-8, 1999. [PUBMED Abstract]
Albores-Saavedra J, Batich K, Chable-Montero F, et al.: Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol 37 (1): 20-7, 2010. [PUBMED Abstract]
Heath M, Jaimes N, Lemos B, et al.: Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol 58 (3): 375-81, 2008. [PUBMED Abstract]
Paulson KG, Park SY, Vandeven NA, et al.: Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J Am Acad Dermatol 78 (3): 457-463.e2, 2018. [PUBMED Abstract]
Ma JE, Brewer JD: Merkel cell carcinoma in immunosuppressed patients. Cancers (Basel) 6 (3): 1328-50, 2014. [PUBMED Abstract]
Howard RA, Dores GM, Curtis RE, et al.: Merkel cell carcinoma and multiple primary cancers. Cancer Epidemiol Biomarkers Prev 15 (8): 1545-9, 2006. [PUBMED Abstract]
Bzhalava D, Bray F, Storm H, et al.: Risk of second cancers after the diagnosis of Merkel cell carcinoma in Scandinavia. Br J Cancer 104 (1): 178-80, 2011. [PUBMED Abstract]
Lunder EJ, Stern RS: Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet A radiation. N Engl J Med 339 (17): 1247-8, 1998. [PUBMED Abstract]
Feng H, Shuda M, Chang Y, et al.: Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319 (5866): 1096-100, 2008. [PUBMED Abstract]
Garneski KM, Warcola AH, Feng Q, et al.: Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J Invest Dermatol 129 (1): 246-8, 2009. [PUBMED Abstract]
Becker JC, Houben R, Ugurel S, et al.: MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J Invest Dermatol 129 (1): 248-50, 2009. [PUBMED Abstract]
Kassem A, Schöpflin A, Diaz C, et al.: Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res 68 (13): 5009-13, 2008. [PUBMED Abstract]
Paik JY, Hall G, Clarkson A, et al.: Immunohistochemistry for Merkel cell polyomavirus is highly specific but not sensitive for the diagnosis of Merkel cell carcinoma in the Australian population. Hum Pathol 42 (10): 1385-90, 2011. [PUBMED Abstract]
Andres C, Belloni B, Puchta U, et al.: Prevalence of MCPyV in Merkel cell carcinoma and non-MCC tumors. J Cutan Pathol 37 (1): 28-34, 2010. [PUBMED Abstract]
Kassem A, Technau K, Kurz AK, et al.: Merkel cell polyomavirus sequences are frequently detected in nonmelanoma skin cancer of immunosuppressed patients. Int J Cancer 125 (2): 356-61, 2009. [PUBMED Abstract]
Foulongne V, Dereure O, Kluger N, et al.: Merkel cell polyomavirus DNA detection in lesional and nonlesional skin from patients with Merkel cell carcinoma or other skin diseases. Br J Dermatol 162 (1): 59-63, 2010. [PUBMED Abstract]
DeCaprio JA: Does detection of Merkel cell polyomavirus in Merkel cell carcinoma provide prognostic information? J Natl Cancer Inst 101 (13): 905-7, 2009. [PUBMED Abstract]
Laude HC, Jonchère B, Maubec E, et al.: Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog 6 (8): , 2010. [PUBMED Abstract]
Buck CB, Lowy DR: Immune readouts may have prognostic value for the course of merkel cell carcinoma, a virally associated disease. J Clin Oncol 29 (12): 1506-8, 2011. [PUBMED Abstract]
Lemos B, Nghiem P: Merkel cell carcinoma: more deaths but still no pathway to blame. J Invest Dermatol 127 (9): 2100-3, 2007. [PUBMED Abstract]
Seattle Cancer Care Alliance: Merkel Cell Carcinoma Information for Patients and Their Physicians: Clinical Photos/Images. Seattle, Wa: Seattle Cancer Care Alliance Skin Oncology Clinic, 2009. Available online. Last accessed May 9, 2025.
Gupta SG, Wang LC, Peñas PF, et al.: Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: The Dana-Farber experience and meta-analysis of the literature. Arch Dermatol 142 (6): 685-90, 2006. [PUBMED Abstract]
Anderson SE, Beer KT, Banic A, et al.: MRI of merkel cell carcinoma: histologic correlation and review of the literature. AJR Am J Roentgenol 185 (6): 1441-8, 2005. [PUBMED Abstract]
Iagaru A, Quon A, McDougall IR, et al.: Merkel cell carcinoma: Is there a role for 2-deoxy-2-[f-18]fluoro-D-glucose-positron emission tomography/computed tomography? Mol Imaging Biol 8 (4): 212-7, 2006 Jul-Aug. [PUBMED Abstract]
Belhocine T, Pierard GE, Frühling J, et al.: Clinical added-value of 18FDG PET in neuroendocrine-merkel cell carcinoma. Oncol Rep 16 (2): 347-52, 2006. [PUBMED Abstract]
Missotten GS, de Wolff-Rouendaal D, de Keizer RJ: Merkel cell carcinoma of the eyelid review of the literature and report of patients with Merkel cell carcinoma showing spontaneous regression. Ophthalmology 115 (1): 195-201, 2008. [PUBMED Abstract]
Richetta AG, Mancini M, Torroni A, et al.: Total spontaneous regression of advanced merkel cell carcinoma after biopsy: review and a new case. Dermatol Surg 34 (6): 815-22, 2008. [PUBMED Abstract]
Allen PJ, Bowne WB, Jaques DP, et al.: Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol 23 (10): 2300-9, 2005. [PUBMED Abstract]
Stokes JB, Graw KS, Dengel LT, et al.: Patients with Merkel cell carcinoma tumors < or = 1.0 cm in diameter are unlikely to harbor regional lymph node metastasis. J Clin Oncol 27 (23): 3772-7, 2009. [PUBMED Abstract]
Jabbour J, Cumming R, Scolyer RA, et al.: Merkel cell carcinoma: assessing the effect of wide local excision, lymph node dissection, and radiotherapy on recurrence and survival in early-stage disease–results from a review of 82 consecutive cases diagnosed between 1992 and 2004. Ann Surg Oncol 14 (6): 1943-52, 2007. [PUBMED Abstract]
Henness S, Vereecken P: Management of Merkel tumours: an evidence-based review. Curr Opin Oncol 20 (3): 280-6, 2008. [PUBMED Abstract]
Skelton HG, Smith KJ, Hitchcock CL, et al.: Merkel cell carcinoma: analysis of clinical, histologic, and immunohistologic features of 132 cases with relation to survival. J Am Acad Dermatol 37 (5 Pt 1): 734-9, 1997. [PUBMED Abstract]
Sandel HD, Day T, Richardson MS, et al.: Merkel cell carcinoma: does tumor size or depth of invasion correlate with recurrence, metastasis, or patient survival? Laryngoscope 116 (5): 791-5, 2006. [PUBMED Abstract]
Llombart B, Monteagudo C, López-Guerrero JA, et al.: Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 46 (6): 622-34, 2005. [PUBMED Abstract]
Senchenkov A, Barnes SA, Moran SL: Predictors of survival and recurrence in the surgical treatment of merkel cell carcinoma of the extremities. J Surg Oncol 95 (3): 229-34, 2007. [PUBMED Abstract]
Goldberg SR, Neifeld JP, Frable WJ: Prognostic value of tumor thickness in patients with Merkel cell carcinoma. J Surg Oncol 95 (8): 618-22, 2007. [PUBMED Abstract]
Heath ML, Nghiem P: Merkel cell carcinoma: if no breslow, then what? J Surg Oncol 95 (8): 614-5, 2007. [PUBMED Abstract]
Tai P: Merkel cell cancer: update on biology and treatment. Curr Opin Oncol 20 (2): 196-200, 2008. [PUBMED Abstract]
Andea AA, Coit DG, Amin B, et al.: Merkel cell carcinoma: histologic features and prognosis. Cancer 113 (9): 2549-58, 2008. [PUBMED Abstract]
Paulson KG, Iyer JG, Tegeder AR, et al.: Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol 29 (12): 1539-46, 2011. [PUBMED Abstract]
Fields RC, Busam KJ, Chou JF, et al.: Recurrence and survival in patients undergoing sentinel lymph node biopsy for merkel cell carcinoma: analysis of 153 patients from a single institution. Ann Surg Oncol 18 (9): 2529-37, 2011. [PUBMED Abstract]
Sihto H, Kukko H, Koljonen V, et al.: Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst 101 (13): 938-45, 2009. [PUBMED Abstract]
Harms KL, Healy MA, Nghiem P, et al.: Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Ann Surg Oncol 23 (11): 3564-3571, 2016. [PUBMED Abstract]
Iyer JG, Storer BE, Paulson KG, et al.: Relationships among primary tumor size, number of involved nodes, and survival for 8044 cases of Merkel cell carcinoma. J Am Acad Dermatol 70 (4): 637-643, 2014. [PUBMED Abstract]
Schwartz JL, Griffith KA, Lowe L, et al.: Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol 29 (8): 1036-41, 2011. [PUBMED Abstract]
Ko JS, Prieto VG, Elson PJ, et al.: Histological pattern of Merkel cell carcinoma sentinel lymph node metastasis improves stratification of Stage III patients. Mod Pathol 29 (2): 122-30, 2016. [PUBMED Abstract]
Eng TY, Boersma MG, Fuller CD, et al.: Treatment of merkel cell carcinoma. Am J Clin Oncol 27 (5): 510-5, 2004. [PUBMED Abstract]
Cellular Classification of Merkel Cell Carcinoma
Although the exact origin and function of the Merkel cell remains under investigation, it is thought to have features of both epithelial and neuroendocrine origin and arise in cells with touch-sensitivity function (mechanoreceptors).[1–4]
Characteristic histopathological features include dense core cytoplasmic neurosecretory granules on electron microscopy and cytokeratin-20 on immunohistochemistry (see Figure 4).[5]
A panel of immunoreagents (see Figure 4) can distinguish between Merkel cell carcinoma (MCC) and other similar-appearing tumors including neuroendocrine carcinoma of the lung (i.e., small cell carcinoma), lymphoma, peripheral primitive neuroectodermal tumor, metastatic carcinoid tumor, and small cell melanoma.[5]
Histologically, MCC has been classified into three distinct subtypes:[6–9]
Trabecular: classic pattern, large-cell type, high density or granules on ultrasound examination.
Intermediate: solid pattern (most common).
Small cell: diffuse, few high-density granules on ultrasound examination (second most common).
Mixtures of variants are common.[6–8] Although some small retrospective case series have suggested correlations between certain histological features and outcome, the evidence remains uncertain.[10–12]
One group has suggested a list of 12 elements that should be described in pathology reports of resected primary lesions and nine elements to be described in pathology reports of sentinel lymph nodes. The prognostic significance of these elements has not been validated prospectively.[13]
If the following data are recorded for every patient with MCC, any patient can be staged with the existing or new staging system:
Size of primary tumor (maximum dimension pathologically or clinically in centimeters).
Presence/absence of primary tumor invasion into bone, muscle, fascia, or cartilage.
Presence/absence of nodal metastasis.
Method used to ascertain status of nodal involvement (clinical or pathological examination).
Presence/absence of distant metastasis.
The College of American Pathologists has published a protocol for the examination of specimens from patients with MCC of the skin.[14]
Bichakjian CK, Nghiem P, Johnson T, et al.: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp 549-62.
Eng TY, Boersma MG, Fuller CD, et al.: A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol 30 (6): 624-36, 2007. [PUBMED Abstract]
Medina-Franco H, Urist MM, Fiveash J, et al.: Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol 8 (3): 204-8, 2001. [PUBMED Abstract]
Busse PM, Clark JR, Muse VV, et al.: Case records of the Massachusetts General Hospital. Case 19-2008. A 63-year-old HIV-positive man with cutaneous Merkel-cell carcinoma. N Engl J Med 358 (25): 2717-23, 2008. [PUBMED Abstract]
Ratner D, Nelson BR, Brown MD, et al.: Merkel cell carcinoma. J Am Acad Dermatol 29 (2 Pt 1): 143-56, 1993. [PUBMED Abstract]
Gould VE, Moll R, Moll I, et al.: Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest 52 (4): 334-53, 1985. [PUBMED Abstract]
Albores-Saavedra J, Batich K, Chable-Montero F, et al.: Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol 37 (1): 20-7, 2010. [PUBMED Abstract]
Alam M: Management of Merkel cell carcinoma: What we know. Arch Dermatol 142 (6): 771-4, 2006. [PUBMED Abstract]
Heath ML, Nghiem P: Merkel cell carcinoma: if no breslow, then what? J Surg Oncol 95 (8): 614-5, 2007. [PUBMED Abstract]
Andea AA, Coit DG, Amin B, et al.: Merkel cell carcinoma: histologic features and prognosis. Cancer 113 (9): 2549-58, 2008. [PUBMED Abstract]
Bichakjian CK, Lowe L, Lao CD, et al.: Merkel cell carcinoma: critical review with guidelines for multidisciplinary management. Cancer 110 (1): 1-12, 2007. [PUBMED Abstract]
Rao P, Balzer BL, Lemos BD, et al.: Protocol for the examination of specimens from patients with merkel cell carcinoma of the skin. Arch Pathol Lab Med 134 (3): 341-4, 2010. [PUBMED Abstract]
Princess Margaret Hospital/University Health Network, Toronto, Canada
Sydney Head and Neck Cancer Institute/Royal Prince Alfred Hospital, Sydney, Australia
These staging systems are highly inconsistent with each other. Stage III disease can mean anything from advanced local disease to nodal disease to distant metastatic disease. Furthermore, all MCC staging systems in use have been based on fewer than 300 patients.
American Joint Committee on Cancer (AJCC) Stage Groupings and TNM Definitions
To address these concerns, a new MCC-specific consensus staging system was developed by the AJCC to define MCC.[7] Before the publication of this new system, the AJCC advocated using the nonmelanoma staging system.
Cancers staged using this staging system include primary cutaneous neuroendocrine carcinoma (MCC).
Clinical Stage Group (cTNM)
Table 3. Definitions of Clinical Stage Group (cTNM) for Stage 0a
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
0
Tis, N0, M0
Tis = In situ primary tumor.
N0 = No regional lymph node metastasis detected on clinical and/or radiologic examination.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
Table 4. Definitions of Clinical Stage Group (cTNM) for Stage Ia
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
I
T1, N0, M0
T1 = Maximum clinical tumor diameter ≤2 cm.
N0 = No regional lymph node metastasis detected on clinical and/or radiologic examination.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
Table 5. Definitions of Clinical Stage Group (cTNM) for Stages IIA and IIBa
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
IIA
T2–3, N0, M0
T2 = Maximum clinical tumor diameter >2 cm but ≤5 cm.
T3 = Maximum clinical tumor diameter >5 cm.
N0 = No regional lymph node metastasis detected on clinical and/or radiologic examination.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
IIB
T4, N0, M0
T4 = Primary tumor invades fascia, muscle, cartilage, or bone.
N0 = No regional lymph node metastasis detected on clinical and/or radiologic examination.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
Table 6. Definitions of Clinical Stage Group (cTNM) for Stage IIIa
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
III
T0–4, N1–3, M0
T0 = No evidence of primary tumor.
Tis = In situ primary tumor.
T1 = Maximum clinical tumor diameter ≤2 cm.
T2 = Maximum clinical tumor diameter >2 but ≤5 cm.
T3 = Maximum clinical tumor diameter >5 cm.
T4 = Primary tumor invades fascia, muscle, cartilage, or bone.
N1 = Metastasis in regional lymph node(s).
N2 = In-transit metastasis (discontinuous from primary tumor; located between primary tumor and draining regional nodal basin, or distal to the primary tumor) without lymph node metastasis.
N3 = In-transit metastasis (discontinuous from primary tumor; located between primary tumor and draining regional nodal basin, or distal to the primary tumor) with lymph node metastasis.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
Table 7. Definitions of Clinical Stage Group (cTNM) for Stage IVa
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; c = clinical.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
IV
T0–4, Any N, M1
T0 = No evidence of primary tumor.
Tis = In situ primary tumor.
T1 = Maximum clinical tumor diameter ≤2 cm.
T2 = Maximum clinical tumor diameter >2 but ≤5 cm.
T3 = Maximum clinical tumor diameter >5 cm.
T4 = Primary tumor invades fascia, muscle, cartilage, or bone.
NX = Regional lymph nodes cannot be clinically assessed (e.g., previously removed for another reason, or because of body habitus).
N0 = No regional lymph node metastasis detected on clinical and/or radiologic examination.
N1 = Metastasis in regional lymph node(s).
N2 = In-transit metastasis (discontinuous from primary tumor; located between primary tumor and draining regional nodal basin, or distal to the primary tumor) without lymph node metastasis.
N3 = In-transit metastasis (discontinuous from primary tumor; located between primary tumor and draining regional nodal basin, or distal to the primary tumor) with lymph node metastasis.
M1 = Distant metastasis detected on clinical and/or radiologic examination.
Pathological Stage Group (pTNM)
Table 8. Definitions of Pathological Stage Group (pTNM) for Stage 0a
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; p = pathological.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
0
Tis, pN0, M0
Tis = In situ primary tumor.
pN0 = No regional lymph node metastasis detected on pathological evaluation.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
Table 9. Definitions of Pathological Stage Group (pTNM) for Stage Ia
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; p = pathological.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
I
T1, pN0, M0
T1 = Maximum clinical tumor diameter ≤2 cm.
pN0 = No regional lymph node metastasis detected on pathological evaluation.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
Table 10. Definitions of Pathological Stage Group (pTNM) for Stages IIA and IIBa
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; p = pathological.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
IIA
T2–3, pN0, M0
T2 = Maximum clinical tumor diameter >2 but ≤5 cm.
T3 = Maximum clinical tumor diameter >5 cm.
pN0 = No regional lymph node metastasis detected on pathological evaluation.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
IIB
T4, pN0, M0
T4 = Primary tumor invades fascia, muscle, cartilage, or bone.
pN0 = No regional lymph node metastasis detected on pathological evaluation.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
Table 11. Definitions of Pathological Stage Group (pTNM) for Stages IIIA and IIIBa
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; p = pathological.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
IIIA
T1–4, pN1a(sn) or pN1a, M0
T1 = Maximum clinical tumor diameter ≤2 cm.
T2 = Maximum clinical tumor diameter >2 cm but ≤5 cm.
T3 = Maximum clinical tumor diameter >5 cm.
T4 = Primary tumor invades fascia, muscle, cartilage, or bone.
pN1a(sn) = Clinically occult regional lymph node metastasis identified only by sentinel lymph node biopsy.
pN2 = In-transit metastasis (discontinuous from primary tumor; located between primary tumor and draining regional nodal basin, or distal to the primary tumor) without lymph node metastasis.
pN3 = In-transit metastasis (discontinuous from primary tumor; located between primary tumor and draining regional nodal basin, or distal to the primary tumor) with lymph node metastasis.
M0 = No distant metastasis detected on clinical and/or radiologic examination.
Table 12. Definitions of Pathological Stage Group (pTNM) Stage IVa
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis; p = pathological.
aReprinted with permission from AJCC: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 549–62.
IV
T0–4, Any pN, pM1
T0 = No evidence of primary tumor.
T1 = Maximum clinical tumor diameter ≤2 cm.
T2 = Maximum clinical tumor diameter >2 cm but ≤5 cm.
T3 = Maximum clinical tumor diameter >5 cm.
T4 = Primary tumor invades fascia, muscle, cartilage, or bone.
pNX = Regional lymph nodes cannot be assessed (e.g., previously removed for another reason or not removed for pathological evaluation).
pN0 = No regional lymph node metastasis detected on pathological evaluation.
pN1 = Metastasis in regional lymph node(s).
–pN1a(sn) = Clinically occult regional lymph node metastasis identified only by sentinel lymph node biopsy.
pN2 = In-transit metastasis (discontinuous from primary tumor; located between primary tumor and draining regional nodal basin, or distal to the primary tumor) without lymph node metastasis.
pN3 = In-transit metastasis (discontinuous from primary tumor; located between primary tumor and draining regional nodal basin, or distal to the primary tumor) with lymph node metastasis.
–pM1a = Metastasis to distant skin, distant subcutaneous tissue, or distant lymph node(s), microscopically confirmed.
–pM1b = Metastasis to lung, microscopically confirmed.
pM1c = Metastasis to all other distant sites, microscopically confirmed.
Before the new AJCC consensus staging system was published, the most recent Memorial Sloan Kettering Cancer Center (MSKCC) four-stage system was favored because it was based on the largest number of patients and was the best validated.[1] The stages in the MSKCC system included:
Stage I: local disease <2 cm.
Stage II: local disease ≥2 cm.
Stage III: regional nodal disease.
Stage IV: distant metastatic disease.
One group has suggested a list of 12 elements that should be described in pathology reports of resected primary lesions and nine elements to be described in pathology reports of sentinel lymph nodes. The prognostic significance of these elements has not been validated prospectively.[8] The 2009 AJCC staging manual also specifies a variety of factors that should be collected prospectively on pathology reports.
References
Andea AA, Coit DG, Amin B, et al.: Merkel cell carcinoma: histologic features and prognosis. Cancer 113 (9): 2549-58, 2008. [PUBMED Abstract]
Yiengpruksawan A, Coit DG, Thaler HT, et al.: Merkel cell carcinoma. Prognosis and management. Arch Surg 126 (12): 1514-9, 1991. [PUBMED Abstract]
Allen PJ, Zhang ZF, Coit DG: Surgical management of Merkel cell carcinoma. Ann Surg 229 (1): 97-105, 1999. [PUBMED Abstract]
Allen PJ, Bowne WB, Jaques DP, et al.: Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol 23 (10): 2300-9, 2005. [PUBMED Abstract]
Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017.
Clark JR, Veness MJ, Gilbert R, et al.: Merkel cell carcinoma of the head and neck: is adjuvant radiotherapy necessary? Head Neck 29 (3): 249-57, 2007. [PUBMED Abstract]
Bichakjian CK, Nghiem P, Johnson T, et al.: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp 549-62.
Bichakjian CK, Lowe L, Lao CD, et al.: Merkel cell carcinoma: critical review with guidelines for multidisciplinary management. Cancer 110 (1): 1-12, 2007. [PUBMED Abstract]
Treatment Option Overview for Merkel Cell Carcinoma
Merkel cell carcinoma (MCC) is an uncommon tumor. Most clinical management recommendations in the literature are based on case series that describe a relatively small number of patients who were not entered on formal clinical trials, evaluated with uniform clinical staging procedures, treated with uniform treatment protocols, or provided with regular, prescribed follow-up. These reports are also confounded by potential selection bias, referral bias, and short follow-up. They are underpowered to detect modest differences in outcome.
In addition, outcomes of patients with American Joint Committee on Cancer stage I and stage II disease are often reported together. In the absence of results from clinical trials with prescribed work-up, treatments, and follow-up, most patients with MCC have been treated using institutional or practitioner preferences that consider the specifics of each case as well as patient preference.
There are two competing philosophies about the most appropriate method of treating MCC. In the first philosophy, MCC is treated like other nonmelanoma skin cancers, with an emphasis on treating local-regional disease with surgery and radiation therapy, as appropriate. In the second philosophy, MCC is treated according to its biological features. This approach makes it analogous to small cell lung cancer, which is assumed to be a systemic disease, and leads to a more routine recommendation of systematic adjuvant chemotherapy.[1]
Surgery for the Primary Lesion
In a review of 18 case series, 279 of 926 patients (30.1%) developed local recurrence during follow-up, excluding those presenting with distant metastatic disease at presentation. These recurrences have been typically attributed to inadequate surgical margins or possibly a lack of adjuvant radiation therapy.[2,3]
Given the propensity of MCC to recur locally (sometimes with satellite lesions and/or in-transit metastases), wide local excision to reduce the risk of local recurrence has been recommended for patients with clinical stage I or stage II disease.
Recommendations about the optimal minimum width and depth of normal tissue margin to be excised around the primary tumor differ among the various retrospective case series, but this question has not been studied systematically.[3–7][Level of evidence C2] No definitive data suggest that extremely wide margins improve overall survival (OS), although some reports suggest that wider margins appear to improve local control.[3][Level of evidence C2] Frozen-section evaluation of margins may be useful, especially when the tumor is in an anatomical site that is not amenable to wide margins.
Some authors have advocated the use of Mohs micrographic surgery as a tissue-sparing technique. The relapse rate has been reported to be similar to or better than that of wide excision, but comparatively few cases have been treated in this manner and none in randomized controlled trials.[7–10][Level of evidence C2]
Regional Lymph Node Surgery
In some case series, local-regional recurrence rates are high when pathological nodal staging is omitted. Surgical nodal staging in clinically negative patients has identified positive nodes in at least 25% to 35% of patients.[4,11,12][Level of evidence C2] In one retrospective series of 213 patients who underwent surgical treatment of the primary tumor and evaluation of the draining nodes, nodal positivity was found in 2 of 54 patients with small tumors (e.g., ≤1.0 cm) and 51 of 159 patients with tumors larger than 1.0 cm.[13][Level of evidence C2]
The role of elective lymph node dissection (ELND) in the absence of clinically positive lymph nodes has not been studied in formal clinical trials. In small case series, ELND has been recommended for larger primary tumors, tumors with more than ten mitoses per high-power field, lymphatic or vascular invasion, and the small-cell histological subtypes.[14–16][Level of evidence C2]
Sentinel lymph node (SLN) biopsy has been suggested as a preferred initial alternative to complete ELND for the proper staging of MCC. SLN biopsy has less morbidity than complete nodal dissection. Furthermore, for MCC sites with indeterminate lymphatic drainage, such as those on the back, SLN biopsy techniques can be used to identify the pertinent lymph node bed(s). If performed, SLN biopsy is done at the time of the wide resection when the local lymphatic channels are still intact.
Several reports have found the use of SLN biopsy techniques in MCC to be reliable and reproducible.[17–20] However, the significance of SLN positivity remains unclear.
One meta-analysis of ten case series found that SLN positivity strongly predicted a high short-term risk of recurrence and that subsequent therapeutic lymph node dissection was effective in preventing short-term regional nodal recurrence.[21]
Another meta-analysis included 12 retrospective case series (only partially overlapping the collection of case series in the previous meta-analysis).[12][Level of evidence C2]
SLN biopsy detected MCC spread in one-third of patients whose tumors would have otherwise been clinically and radiologically understaged.
The recurrence rate was three times higher in patients with a positive SLN biopsy than in those with a negative SLN biopsy (P = .03).
Between 2006 and 2010, a large, retrospective, single-institutional series of 95 patients (with a total of 97 primary tumors) identified a SLN in 93 instances, and nodal tumor was seen in 42 patients. Immunohistochemical techniques were used to assess node positivity. Various models of tumor and patient characteristics were studied to predict node positivity. There was no subgroup of patients predicted to have lower than 15% to 20% likelihood of SLN positivity, suggesting that SLN biopsy may be considered for all curative patients with clinically negative lymph nodes and no distant metastases.[22][Level of evidence C2]
From 1996 to 2010, another retrospective single-institutional study of 153 patients with localized MCC who underwent SLN biopsy analyzed factors associated with SLN positivity. The best predictors of SLN biopsy positivity were tumor size and lymphovascular invasion.[22,23][Level of evidence C2]
In the absence of adequately powered, prospective, randomized clinical trials, the following questions remain:[4,12,21,24][Level of evidence C2]
Should every positive SLN biopsy be followed routinely by completion nodal surgery and/or radiation therapy?
Are outcomes demonstrably improved by routinely adding radiation if lymph node surgery reveals tumor in multiple nodes and/or extracapsular extension and/or lymphovascular invasion?
Should patients with MCCs smaller than 1 cm routinely undergo sentinel lymph node dissection (SLND)?
Should patients with negative or omitted nodal work-up routinely undergo local or local-regional radiation therapy?
Should immunohistochemical staining techniques be used to identify micrometastases in lymph nodes, and is micrometastatic disease in nodes clinically relevant?
Lymph node surgery is primarily used for staging and guiding additional treatment.
Based on a small number of retrospective studies, therapeutic dissection of the regional lymph nodes after a positive SLND appears to minimize, but not totally eliminate, the risk of subsequent regional lymph node recurrence and in-transit metastases.[4,21,24][Level of evidence C2] There are no data from prospective randomized trials demonstrating that definitive regional nodal treatment with surgery improves survival.
Radiation Therapy
Because of the aggressive nature of MCC, its apparent radiosensitivity, and the high incidence of local and regional recurrences (including in-transit metastases after surgery alone to the primary tumor bed), some clinicians have recommended adjuvant radiation therapy to the primary site and nodal basin. Nodal basin radiation in contiguity with radiation to the primary site has been considered, especially for patients with larger tumors, locally unresectable tumors, close or positive excision margins that cannot be improved by additional surgery, and those with positive regional lymph nodes, especially after SLND (stage II).[10,11,14,15,25][Level of evidence C2] Several small retrospective series have shown that radiation therapy plus adequate surgery improves local-regional control compared with surgery alone,[2,5,26–29] whereas other series did not show the same results.[4,8][Level of evidence C2]
In the absence of adequately powered, prospective, randomized clinical trials, the following questions remain:[4,8,9,12,21,24,26,30–34][Level of evidence C2]
Should every positive SLN biopsy be followed routinely by completion nodal surgery and/or radiation therapy?
Are outcomes demonstrably improved by routinely adding radiation only if nodal surgery reveals tumor in multiple lymph nodes and/or extracapsular extension and/or lymphovascular invasion?
Should all or just certain patients with negative or omitted nodal work-up receive local or local-regional radiation routinely?
Because of the small size of these nonrandomized retrospective series, the precise benefit from radiation therapy remains unproven.
When recommended, the radiation dose given has been at least 50 Gy to the surgical bed with margins and to the draining regional lymphatics, delivered in 2 Gy fractions. For patients with unresected tumors or tumors with microscopic evidence of spread beyond resected margins, higher doses of 56 Gy to 65 Gy to the primary site have been recommended.[5,10,11,14,15,27,31,35][Level of evidence C2] These doses have not been studied prospectively in clinical trials.
Local and/or regional control of MCC with radiation therapy alone has been reported in small, highly selected, nonrandomized case series of patients with diverse clinical characteristics.[29,36] Typically, these patients have had inoperable primary tumors and/or nodes or were considered medically inappropriate for surgery.[29,36][Level of evidence C2]
Retrospective Surveillance, Epidemiology, and End Results (SEER) Program data suggest that adding radiation therapy to surgery adds survival value, but the conclusions are complicated by incomplete patient data, no protocol for evaluation and treatment, and potential sampling bias.[32] Prospective randomized clinical trials are required to assess whether combining surgery with radiation therapy affects survival.[33,34][Level of evidence C2]
Immunotherapy
Approximately 70% to 80% of MCC cases in the United States are caused by Merkel cell polyomavirus (MCPyV). Within virus-positive MCCs, the viral oncoproteins (T antigens) are constitutively expressed and promote growth. Furthermore, patients with stimulated immune responses to MCPyV have better disease outcomes, providing a rationale for the use of immunotherapy. Although limited by a lack of randomized trials, several single-agent immune checkpoint inhibitors have shown improved survival and tolerability in patients with advanced MCC, compared with the chemotherapy that was used in historical controls. Therefore, immune checkpoint inhibitors are considered the recommended first-line treatment for most patients. There are ongoing trials to assess the use of immune checkpoint inhibitors in the neoadjuvant and adjuvant setting.
Avelumab
Avelumab is a human anti–programmed death ligand-1 (PD-L1) monoclonal antibody.
Evidence (avelumab):
In a phase II trial (JAVELIN Merkel 200 [NCT02155647]), 88 patients with metastatic MCC received avelumab (10 mg/kg intravenously [IV] every 2 weeks). These patients had previously received chemotherapy.[37]
The objective response rate was 33%, and 11% of patients had a complete response. The median time to response was 6.1 weeks and was irrespective of MCPyV or PD-L1 status. More importantly, responses were durable, with 74% lasting at least 1 year. The median OS was 12.6 months, more than twice the historical median with second-line chemotherapy.
Based on the results of this study, the U.S. Food and Drug Administration (FDA) approved avelumab in 2017 for the treatment of patients with metastatic MCC, regardless of previous chemotherapy administration.[37–39]
Avelumab was also studied in the first-line setting in a single-arm phase II trial of 116 patients.[40]
After a median follow-up of 21.2 months, 30.2% (95% confidence interval [CI], 22.0%–39.4%) of patients had a durable response (>6 months). The objective response rate was 39.7%, and the median OS was 20.3 months (95% CI, 12.4–not estimable).[40][Level of evidence C3]
Exploratory analyses found that responses were higher in patients with PD-L1 expression and in those negative for MCPyV.
Pembrolizumab
Pembrolizumab is a humanized IgG4 anti–programmed cell death-1 (PD-1) monoclonal antibody.
Evidence (pembrolizumab):
Pembrolizumab was studied in a phase II trial (NCT02267603) of first-line systemic treatment for patients with unresectable stage IIIB or stage IV MCC. A total of 50 patients received therapy.[41][Level of evidence C3]
The objective response rate was 58% (95% CI, 43.2%–71.8%); 30% of patients had a complete response and 28% had a partial response. While the response rate was similar to historical rates with first-line chemotherapy, responses to pembrolizumab were more durable. The median progression-free survival (PFS) was 16.8 months (95% CI, 4.6–43.4), and the 3-year OS rate was 59.4%.
The FDA approved pembrolizumab in 2018.
Retifanlimab
Retifanlimab is an anti–PD-1 monoclonal antibody.
Evidence (retifanlimab):
Retifanlimab was evaluated in a phase II trial (POD1UM-201 [NCT03599713]), published in abstract form, of 101 patients with previously untreated advanced MCC. Patients received 500mg intravenously (IV) every 4 weeks.[42,43]
Preliminary results after a median follow up 17.6 months demonstrated a complete response rate of 16.8%, a partial response rate of 38.6%, and an overall response rate of 53.5% (95% CI, 43.3%–63.5%).[42,43][Level of evidence C3]
The median PFS was 12.7 months (95% CI, 7.3–24.9) and the median duration of response was 25.3 months (95% CI, 14.2–not estimable). The median OS was not yet reached.
Based on the results of this trial, the FDA granted accelerated approval to retifanlimab in 2023.
Nivolumab
Nivolumab is an anti–PD-1 monoclonal antibody.
Evidence (nivolumab):
Nivolumab was studied in a phase I/II trial (CheckMate 358 [NCT02488759]), published in abstract form, in patients with virus-associated cancers, including MCC. Patients may have received up to two prior lines of therapy. Patients with metastatic MCC were eligible regardless of MCPyV status or previous chemotherapy.[44,45]
Data from 25 patients with metastatic disease demonstrated an overall response rate of 60% with a median treatment duration of 15.8 months. Preliminary results reported an ongoing response rate of 87%, with responses in 13 of 15 patients at last follow-up (median follow-up, 6 months).[44,45][Level of evidence C3]
This trial added a second cohort to investigate nivolumab plus ipilimumab (1 mg/kg) in patients with metastatic MCC.
Nivolumab is not approved by the FDA for the treatment of MCC.
Chemotherapy
A variety of chemotherapy regimens have been used for patients with MCC in the adjuvant, advanced, and recurrent therapy settings.[5,34,46,47] [Level of evidence C2] No phase III clinical trials have been conducted to demonstrate that adjuvant chemotherapy produces improvements in OS. However, some clinicians recommend its use in most cases because of the following factors:
A biological analogy is made between MCC and the histologically similar small cell carcinoma of the lung, which is considered a systemic disease.
The risk of metastases and progression is high with MCC.
Good initial clinical response rates have been noted with some chemotherapy regimens.
When possible, patients can be encouraged to participate in clinical trials.
From 1997 to 2001, the Trans-Tasman Radiation Oncology Group performed a phase II evaluation of 53 patients with high-risk, local-regional MCC. High risk was defined as recurrence after initial therapy, involved lymph nodes, primary tumor larger than 1 cm, gross residual disease after surgery, or occult primary tumor with positive lymph nodes. Therapy included local-regional radiation therapy (50 Gy in 25 fractions), synchronous carboplatin (area under the curve [AUC], 4.5), and IV etoposide (89 mg/m2 on days 1–3 in weeks 1, 4, 7, and 10). Surgery was not standardized for either the primary tumor or the lymph nodes, and 12 patients had close margins, positive margins, or gross residual disease. Twenty-eight patients had undissected nodal beds, and the remainder had a variety of nodal surgeries. With a median follow-up of 48 months, the 3-year OS rate was 76%, the rate of local-regional control was 75%, and the rate of distant control was 76%. Radiation reactions in the skin and febrile neutropenia were significant clinical acute toxicities. Because of the heterogeneity of the population and the nonstandardized surgery, it is difficult to infer a clear treatment benefit from the chemotherapy.[48][Level of evidence C1]
In a subsequent report, the same investigators evaluated a subset of these protocol patients (n = 40, after excluding patients with unknown primary tumors). These patients were compared with 61 historical controls who received no chemotherapy, were treated at the same institutions, were diagnosed before 1997, and had no routine imaging staging studies. Radiation therapy was given to 50 patients. There was no significant survival benefit seen for chemotherapy patients.[49]
In a subsequent pilot clinical trial of 18 patients from 2004 to 2006, the same investigators attempted to reduce the skin and hematologic toxicity seen in Study 96-07. Carboplatin (AUC, 2) was administered weekly during radiation therapy beginning on day 1 for a maximum of five doses, followed by three cycles of carboplatin (AUC, 4.5; and IV etoposide 80 mg/m2 on days 1–3 beginning 3 weeks after radiation and repeated every 3 weeks for three cycles). The radiation therapy was similar to that in the earlier trial.[48] Results suggested a decrease in hematologic and skin toxicity.[50]
Use of chemotherapy has also been reported in selected patients with locally advanced and metastatic disease. In one retrospective study of 107 patients, 57% of patients with metastatic disease and 69% with locally advanced disease responded to initial chemotherapy. The median OS was 9 months for patients with metastatic disease and 24 months for patients with locally advanced disease. At 3 years, the OS rate was projected to be 17% for those with metastatic disease and 35% for those with locally advanced disease. Toxicity was significant, however, and without clear benefit, particularly in older patients.[51][Level of evidence C2]
Follow-Up
The most appropriate follow-up techniques and frequency for patients treated for MCC have not been prospectively studied. Because of the propensity for local and regional recurrence, clinicians should perform at least a thorough physical examination of the site of initial disease and the regional lymph nodes. Imaging studies may be ordered to evaluate signs and symptoms of concern, or they may be performed to identify distant metastases early. However, there are no data suggesting that early detection and treatment of new distant metastases results in improved survival.
In one series of 237 patients presenting with local or regional disease, the median time-to-recurrence was 9 months (range, 2–70 months). Ninety-one percent of recurrences occurred within 2 years of diagnosis.[4] It has been suggested that the intensity of follow-up can be gradually diminished after 2 to 3 years because most recurrences are likely to have already occurred.[4]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Busse PM, Clark JR, Muse VV, et al.: Case records of the Massachusetts General Hospital. Case 19-2008. A 63-year-old HIV-positive man with cutaneous Merkel-cell carcinoma. N Engl J Med 358 (25): 2717-23, 2008. [PUBMED Abstract]
Medina-Franco H, Urist MM, Fiveash J, et al.: Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol 8 (3): 204-8, 2001. [PUBMED Abstract]
Nghiem P, James N: Merkel cell carcinoma. In: Wolff K, Goldsmith LA, Katz SI, et al., eds.: Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill , 2008, pp 1087-94.
Allen PJ, Bowne WB, Jaques DP, et al.: Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol 23 (10): 2300-9, 2005. [PUBMED Abstract]
Senchenkov A, Barnes SA, Moran SL: Predictors of survival and recurrence in the surgical treatment of merkel cell carcinoma of the extremities. J Surg Oncol 95 (3): 229-34, 2007. [PUBMED Abstract]
Boyer JD, Zitelli JA, Brodland DG, et al.: Local control of primary Merkel cell carcinoma: review of 45 cases treated with Mohs micrographic surgery with and without adjuvant radiation. J Am Acad Dermatol 47 (6): 885-92, 2002. [PUBMED Abstract]
Wilson LD, Gruber SB: Merkel cell carcinoma and the controversial role of adjuvant radiation therapy: clinical choices in the absence of statistical evidence. J Am Acad Dermatol 50 (3): 435-7; discussion 437-8, 2004. [PUBMED Abstract]
Gollard R, Weber R, Kosty MP, et al.: Merkel cell carcinoma: review of 22 cases with surgical, pathologic, and therapeutic considerations. Cancer 88 (8): 1842-51, 2000. [PUBMED Abstract]
Eng TY, Boersma MG, Fuller CD, et al.: A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol 30 (6): 624-36, 2007. [PUBMED Abstract]
Gupta SG, Wang LC, Peñas PF, et al.: Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: The Dana-Farber experience and meta-analysis of the literature. Arch Dermatol 142 (6): 685-90, 2006. [PUBMED Abstract]
Stokes JB, Graw KS, Dengel LT, et al.: Patients with Merkel cell carcinoma tumors < or = 1.0 cm in diameter are unlikely to harbor regional lymph node metastasis. J Clin Oncol 27 (23): 3772-7, 2009. [PUBMED Abstract]
Ratner D, Nelson BR, Brown MD, et al.: Merkel cell carcinoma. J Am Acad Dermatol 29 (2 Pt 1): 143-56, 1993. [PUBMED Abstract]
Yiengpruksawan A, Coit DG, Thaler HT, et al.: Merkel cell carcinoma. Prognosis and management. Arch Surg 126 (12): 1514-9, 1991. [PUBMED Abstract]
Messina JL, Reintgen DS, Cruse CW, et al.: Selective lymphadenectomy in patients with Merkel cell (cutaneous neuroendocrine) carcinoma. Ann Surg Oncol 4 (5): 389-95, 1997 Jul-Aug. [PUBMED Abstract]
Hill AD, Brady MS, Coit DG: Intraoperative lymphatic mapping and sentinel lymph node biopsy for Merkel cell carcinoma. Br J Surg 86 (4): 518-21, 1999. [PUBMED Abstract]
Wasserberg N, Schachter J, Fenig E, et al.: Applicability of the sentinel node technique to Merkel cell carcinoma. Dermatol Surg 26 (2): 138-41, 2000. [PUBMED Abstract]
Rodrigues LK, Leong SP, Kashani-Sabet M, et al.: Early experience with sentinel lymph node mapping for Merkel cell carcinoma. J Am Acad Dermatol 45 (2): 303-8, 2001. [PUBMED Abstract]
Mehrany K, Otley CC, Weenig RH, et al.: A meta-analysis of the prognostic significance of sentinel lymph node status in Merkel cell carcinoma. Dermatol Surg 28 (2): 113-7; discussion 117, 2002. [PUBMED Abstract]
Schwartz JL, Griffith KA, Lowe L, et al.: Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol 29 (8): 1036-41, 2011. [PUBMED Abstract]
Fields RC, Busam KJ, Chou JF, et al.: Recurrence and survival in patients undergoing sentinel lymph node biopsy for merkel cell carcinoma: analysis of 153 patients from a single institution. Ann Surg Oncol 18 (9): 2529-37, 2011. [PUBMED Abstract]
Maza S, Trefzer U, Hofmann M, et al.: Impact of sentinel lymph node biopsy in patients with Merkel cell carcinoma: results of a prospective study and review of the literature. Eur J Nucl Med Mol Imaging 33 (4): 433-40, 2006. [PUBMED Abstract]
Goepfert H, Remmler D, Silva E, et al.: Merkel cell carcinoma (endocrine carcinoma of the skin) of the head and neck. Arch Otolaryngol 110 (11): 707-12, 1984. [PUBMED Abstract]
Lewis KG, Weinstock MA, Weaver AL, et al.: Adjuvant local irradiation for Merkel cell carcinoma. Arch Dermatol 142 (6): 693-700, 2006. [PUBMED Abstract]
Jabbour J, Cumming R, Scolyer RA, et al.: Merkel cell carcinoma: assessing the effect of wide local excision, lymph node dissection, and radiotherapy on recurrence and survival in early-stage disease–results from a review of 82 consecutive cases diagnosed between 1992 and 2004. Ann Surg Oncol 14 (6): 1943-52, 2007. [PUBMED Abstract]
Veness M, Foote M, Gebski V, et al.: The role of radiotherapy alone in patients with merkel cell carcinoma: reporting the Australian experience of 43 patients. Int J Radiat Oncol Biol Phys 78 (3): 703-9, 2010. [PUBMED Abstract]
Meeuwissen JA, Bourne RG, Kearsley JH: The importance of postoperative radiation therapy in the treatment of Merkel cell carcinoma. Int J Radiat Oncol Biol Phys 31 (2): 325-31, 1995. [PUBMED Abstract]
Marks ME, Kim RY, Salter MM: Radiotherapy as an adjunct in the management of Merkel cell carcinoma. Cancer 65 (1): 60-4, 1990. [PUBMED Abstract]
Mojica P, Smith D, Ellenhorn JD: Adjuvant radiation therapy is associated with improved survival in Merkel cell carcinoma of the skin. J Clin Oncol 25 (9): 1043-7, 2007. [PUBMED Abstract]
Housman DM, Decker RH, Wilson LD: Regarding adjuvant radiation therapy in merkel cell carcinoma: selection bias and its affect on overall survival. J Clin Oncol 25 (28): 4503-4; author reply 4504-5, 2007. [PUBMED Abstract]
Garneski KM, Nghiem P: Merkel cell carcinoma adjuvant therapy: current data support radiation but not chemotherapy. J Am Acad Dermatol 57 (1): 166-9, 2007. [PUBMED Abstract]
Foote M, Harvey J, Porceddu S, et al.: Effect of radiotherapy dose and volume on relapse in Merkel cell cancer of the skin. Int J Radiat Oncol Biol Phys 77 (3): 677-84, 2010. [PUBMED Abstract]
Fang LC, Lemos B, Douglas J, et al.: Radiation monotherapy as regional treatment for lymph node-positive Merkel cell carcinoma. Cancer 116 (7): 1783-90, 2010. [PUBMED Abstract]
Kaufman HL, Russell JS, Hamid O, et al.: Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J Immunother Cancer 6 (1): 7, 2018. [PUBMED Abstract]
Becker JC, Lorenz E, Ugurel S, et al.: Evaluation of real-world treatment outcomes in patients with distant metastatic Merkel cell carcinoma following second-line chemotherapy in Europe. Oncotarget 8 (45): 79731-79741, 2017. [PUBMED Abstract]
Cowey CL, Mahnke L, Espirito J, et al.: Real-world treatment outcomes in patients with metastatic Merkel cell carcinoma treated with chemotherapy in the USA. Future Oncol 13 (19): 1699-1710, 2017. [PUBMED Abstract]
D’Angelo SP, Lebbé C, Mortier L, et al.: First-line avelumab in a cohort of 116 patients with metastatic Merkel cell carcinoma (JAVELIN Merkel 200): primary and biomarker analyses of a phase II study. J Immunother Cancer 9 (7): , 2021. [PUBMED Abstract]
Nghiem PT, Bhatia S, Lipson EJ, et al.: PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med 374 (26): 2542-52, 2016. [PUBMED Abstract]
Grignani G, Rutkowski P, Lebbe C, et al.: Updated results from POD1UM-201: A phase II study of retifanlimab in patients with advanced or metastatic Merkel cell carcinoma (MCC). [Abstract] Ann Oncol 34 (Suppl 2): A-1146P, S686, 2023.
Grignani G, Rutkowski P, Lebbé C, et al.: Updated results from POD1UM-201: A phase 2 study of retifanlimab in patients with advanced or metastatic Merkel cell carcinoma. In: Grignani G, Rutkowski P, Lebbe C, et al.: Updated results from POD1UM-201: A phase II study of retifanlimab in patients with advanced or metastatic Merkel cell carcinoma (MCC). 34 (Suppl 2): A-1146P, S686, 2023, Poster. Available online. Last accessed May 9, 2025.
Bhatia S, Topalian SL, Sharfman WH, et al.: Non-comparative, open-label, international, multicenter phase I/II study of nivolumab (NIVO) ± ipilimumab (IPI) in patients (pts) with recurrent/metastatic merkel cell carcinoma (MCC) (CheckMate 358). [Abstract] J Clin Oncol 41 (Suppl 16): A-9506, 2023.
Topalian SL, Bhatia S, Hollebecque A, et al.: Abstract CT074: Non-comparative, open-label, multiple cohort, phase 1/2 study to evaluate nivolumab (NIVO) in patients with virus-associated tumors (CheckMate 358): efficacy and safety in Merkel cell carcinoma (MCC). [Abstract] Cancer Res 77 (13 Suppl): A-CT074, 2017.
Tai PT, Yu E, Winquist E, et al.: Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: case series and review of 204 cases. J Clin Oncol 18 (12): 2493-9, 2000. [PUBMED Abstract]
Henness S, Vereecken P: Management of Merkel tumours: an evidence-based review. Curr Opin Oncol 20 (3): 280-6, 2008. [PUBMED Abstract]
Poulsen M, Rischin D, Walpole E, et al.: High-risk Merkel cell carcinoma of the skin treated with synchronous carboplatin/etoposide and radiation: a Trans-Tasman Radiation Oncology Group Study–TROG 96:07. J Clin Oncol 21 (23): 4371-6, 2003. [PUBMED Abstract]
Poulsen MG, Rischin D, Porter I, et al.: Does chemotherapy improve survival in high-risk stage I and II Merkel cell carcinoma of the skin? Int J Radiat Oncol Biol Phys 64 (1): 114-9, 2006. [PUBMED Abstract]
Poulsen M, Walpole E, Harvey J, et al.: Weekly carboplatin reduces toxicity during synchronous chemoradiotherapy for Merkel cell carcinoma of skin. Int J Radiat Oncol Biol Phys 72 (4): 1070-4, 2008. [PUBMED Abstract]
Voog E, Biron P, Martin JP, et al.: Chemotherapy for patients with locally advanced or metastatic Merkel cell carcinoma. Cancer 85 (12): 2589-95, 1999. [PUBMED Abstract]
Treatment of Stage I and II Merkel Cell Carcinoma
Stage I and II Merkel cell carcinoma (MCC) include patients with local disease only.
Excision with 1 cm to 2 cm margins and radiation therapy are the mainstays of management for primary MCC tumors. Adjuvant radiation therapy to the primary tumor site is often recommended. However, the morbidity of radiation may be avoided and low local recurrence rates maintained, as shown in a subset of patients with small low-risk lesions (i.e., tumors <2 cm without other adverse prognostic factors).[1]
Because of the risk of occult nodal disease, sentinel lymph node (SLN) biopsy is recommended for patients without clinically detectable metastatic disease.[2] Any size of metastatic deposit is currently considered positive with regard to regional lymph node (N) staging; therefore, immunohistochemistry is routinely used to improve detection of micrometastases in SLN.[3,4]
Treatment Options for Stage I and II Merkel Cell Carcinoma
Margin-negative local excision, attempting to maintain function.
Surgical nodal evaluation, typically by SLN procedure initially, may be considered for patients with significant risk of nodal disease. Completion of the nodal dissection may be considered if positive lymph nodes are found, which would upstage the patient’s cancer to stage III.
Local radiation therapy may be considered if there is concern about the primary tumor excision margins. Regional radiation therapy may be considered if the nodal staging procedure is incomplete or omitted. For sites where the location of primary regional lymph nodes may be uncertain (e.g., mid-back), regional-node field selection is problematic.
Enrollment in clinical trials is encouraged.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Frohm ML, Griffith KA, Harms KL, et al.: Recurrence and Survival in Patients With Merkel Cell Carcinoma Undergoing Surgery Without Adjuvant Radiation Therapy to the Primary Site. JAMA Dermatol 152 (9): 1001-7, 2016. [PUBMED Abstract]
Margin-negative local excision, attempting to maintain function.
Sentinel lymph node procedure, possibly followed by more definitive regional node surgery if positive node(s) are found.
Local and regional lymph node radiation, especially if there is concern about the adequacy of the primary tumor excision margin or the risk of local-regional recurrence following the nodal surgery (e.g., multiple primary nodes, extracapsular extension, lymphovascular invasion, and evidence of in-transit metastases).
Systemic chemotherapy and immunotherapy have been given to patients with the highest risk of recurrence, but existing data have not proven a clinical survival benefit.
Enrollment in clinical trials is encouraged.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Treatment of Stage IV Merkel Cell Carcinoma
Stage IV Merkel cell carcinoma (MCC) includes patients with distant metastases.
Single-agent immunotherapy is the preferred initial systemic therapy for patients with metastatic disease. Chemotherapy may be considered for patients with stage IV disease who have a good performance status and are either ineligible for or have disease progression while receiving immunotherapy. Although responses have been seen with various regimens, evidence is lacking that chemotherapy results in permanent disease control or long-term survival.
If chemotherapy is not considered an appropriate option, patients with stage IV disease may consider surgery and/or radiation therapy for local or regional palliation.
Treatment Options for Stage IV Merkel Cell Carcinoma
Palliation with chemotherapy and/or surgery and/or radiation therapy as clinically appropriate.
Enrollment in clinical trials is strongly encouraged.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Treatment of Recurrent Merkel Cell Carcinoma
Merkel cell carcinoma (MCC) is a rare tumor. Recommendations and outcomes of various treatments for patients with MCC are included in many large case series [1–3][Level of evidence C2] and several single-arm phase II clinical trials.[4][Level of evidence C1] Treatment is usually individualized based on patient preference and the specifics of each case. Patients should strongly consider enrolling in clinical trials.
Treatment Options for Recurrent Merkel Cell Carcinoma
Local recurrence
Treatment options for patients with local recurrence include wider local surgery if possible, followed by radiation therapy, if not previously given.
Regional lymph node dissection (RLND) can also be considered if regional draining nodes have not been previously removed.
Because of the poor prognosis after recurrence, systemic therapy can also be considered, although there is no evidence that it improves survival.
Nodal recurrence
Treatment options for patients with only regional nodal recurrence include RLND and adjuvant radiation therapy if the regional draining nodes have not been previously treated. Because of the poor prognosis after recurrence, consideration can also be given to systemic therapy, although there is no evidence that it improves survival.
Distant recurrence
For patients with distant disease, several single-agent immunotherapies have elicited durable responses and are the recommended first-line systemic therapy option for most patients. Among these agents, avelumab and pembrolizumab have longer-term follow-up data published. These agents are approved by the U.S. Food and Drug Administration (FDA).[5–7][Level of evidence C3] Retifanlimab has also received accelerated approval from the FDA, pending further follow-up.[8][Level of evidence C3]
Chemotherapy is an option for patients who are either ineligible for or have disease progression while receiving immunotherapy, or in select patients who may benefit from a more rapid response.[1–4,9,10][Level of evidence C2] Although responses with chemotherapy have been reported in selected patients with locally advanced and metastatic disease, toxicity has been significant and durability is limited. When appropriate, radiation therapy and/or surgery may be offered as palliation to sites of recurrence, particularly if chemotherapy is not considered an option.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Henness S, Vereecken P: Management of Merkel tumours: an evidence-based review. Curr Opin Oncol 20 (3): 280-6, 2008. [PUBMED Abstract]
Voog E, Biron P, Martin JP, et al.: Chemotherapy for patients with locally advanced or metastatic Merkel cell carcinoma. Cancer 85 (12): 2589-95, 1999. [PUBMED Abstract]
Poulsen M, Rischin D, Walpole E, et al.: High-risk Merkel cell carcinoma of the skin treated with synchronous carboplatin/etoposide and radiation: a Trans-Tasman Radiation Oncology Group Study–TROG 96:07. J Clin Oncol 21 (23): 4371-6, 2003. [PUBMED Abstract]
Kaufman HL, Russell JS, Hamid O, et al.: Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J Immunother Cancer 6 (1): 7, 2018. [PUBMED Abstract]
D’Angelo SP, Lebbé C, Mortier L, et al.: First-line avelumab in a cohort of 116 patients with metastatic Merkel cell carcinoma (JAVELIN Merkel 200): primary and biomarker analyses of a phase II study. J Immunother Cancer 9 (7): , 2021. [PUBMED Abstract]
Nghiem PT, Bhatia S, Lipson EJ, et al.: PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med 374 (26): 2542-52, 2016. [PUBMED Abstract]
Grignani G, Rutkowski P, Lebbe C, et al.: Updated results from POD1UM-201: A phase II study of retifanlimab in patients with advanced or metastatic Merkel cell carcinoma (MCC). [Abstract] Ann Oncol 34 (Suppl 2): A-1146P, S686, 2023.
Eng TY, Boersma MG, Fuller CD, et al.: A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol 30 (6): 624-36, 2007. [PUBMED Abstract]
Tai PT, Yu E, Winquist E, et al.: Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: case series and review of 204 cases. J Clin Oncol 18 (12): 2493-9, 2000. [PUBMED Abstract]
Latest Updates to This Summary (05/09/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of Merkel cell carcinoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Merkel Cell Carcinoma Treatment is:
Shaheer A. Khan, DO (Columbia University Irving Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Merkel Cell Carcinoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/skin/hp/merkel-cell-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 20943647]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
The most common site for neuroendocrine (carcinoid) tumors is the appendix. In a single-institution retrospective review of 45 cases of neuroendocrine (carcinoid) tumors in children and adolescents between 2003 and 2016, the appendix was the primary site in 36 patients.[1][Level of evidence C2] No recurrences were observed among the patients with appendiceal primary tumors treated with appendectomy alone, which supports resection of the appendix without hemicolectomy as the procedure of choice.
Most neuroendocrine tumors of the appendix are discovered incidentally at the time of appendectomy. Most of them are small, low-grade, localized tumors.[2–4]
Treatment of Gastrointestinal Neuroendocrine Tumors of the Appendix
Treatment options for neuroendocrine tumors of the appendix include the following:
Appendectomy.
In adults, it has been accepted practice to remove the entire right colon in patients with large neuroendocrine tumors of the appendix (>2 cm in diameter) or with tumors that have spread to the lymph nodes.[5–8]
In children and adolescents, however, study results suggest that appendectomy alone is sufficient treatment for appendiceal neuroendocrine tumors, regardless of size, position, histology, or nodal or mesenteric involvement. Right hemicolectomy is unnecessary in children. Routine follow-up imaging and biological studies are not beneficial.[5,8–10]
Evidence (appendectomy alone):
The Italian Tumori Rari in Etá Pediatrica project performed a prospective registry study that evaluated 113 pediatric patients with appendiceal neuroendocrine tumors.[9][Level of evidence C1] Primary re-excision was not recommended for completely excised tumors smaller than 2 cm, except for microscopic/macroscopic residual tumor on the margins of the appendix. In these cases, cecum resection and pericecal node biopsy was recommended. Decisions about tumors larger than 2 cm were made at the discretion of the primary physicians. However, physicians were discouraged from performing right hemicolectomy unless margins were positive. Of the 113 study participants, 108 had tumors smaller than 2 cm. Thirty-five patients had extension of tumor beyond the appendiceal wall. Five tumors invaded the serosa, and 28 tumors invaded the periappendiceal fat. Margins were clear in 111 of 113 patients.
At 41 months of follow-up, all 113 patients were alive.
The five patients with tumors larger than 2 cm did well.
One patient had resection of the cecum; no residual tumor was found.
One patient had a right hemicolectomy (tumor was <2 cm with clear margins, but an octreotide scan was possibly positive), and no tumor was found.
The study concluded that appendectomy alone should be considered curative for most pediatric patients with appendiceal neuroendocrine tumors. The procedure of choice is a resection of the appendix without hemicolectomy.
A French multicenter study of children younger than 18 years with neuroendocrine tumors of the appendix was carried out by surveying pediatric surgeons from 1988 to 2012. A total of 114 patients were identified. Risk factors for secondary right hemicolectomy were tumor extension into the mesoappendix, positive margins, size larger than 2 cm, and high proliferative index. Eighteen patients met the above criteria and were observed.[10]
All patients were alive and had no disease at follow-up.
In addition, follow-up radiological studies and biological tests were not helpful.
The investigator’s recommendation was that appendectomy alone is sufficient treatment for neuroendocrine tumors of the appendix.
A systematic review and meta-analysis of 38 studies of appendiceal neuroendocrine tumors identified 958 patients with a mean age at presentation of 11.6 years. Tumor size was 2 cm or smaller in 85% of the cases. Of the 24 papers that reported the status of the resection margin, 97% of tumors had negative margins. Nodal involvement was reported in ten series and was present in 1.4% of cases, with higher rates seen in patients whose tumors were larger than 2 cm (35%). Vascular involvement was seen in 11% of 510 patients, and invasion of the mesoappendix or periappendiceal fat was reported in 29% of 910 patients.[8]
According to the European and American Neuroendocrine Tumor Societies, 189 patients met the criteria for a secondary procedure after initial appendectomy, but only 69 patients underwent a secondary procedure (n = 43, hemicolectomy; n = 2, ileocecectomy; n = 1, cecectomy; n = 2, ileocolectomy; n = 21, not specified).
Of the 120 patients who did not have a secondary procedure, 91 patients had tumors extending to the mesoappendix, 5 patients had vascular invasion, 4 patients had positive margins, 12 patients had tumors 2 cm or larger, 1 patient had a high proliferative index, and 7 patients had positive lymph nodes.
No disease recurrences were reported in patients who had a secondary procedure or in those who were observed.
Preoperative and postoperative imaging was not helpful in managing patients.
References
Degnan AJ, Tocchio S, Kurtom W, et al.: Pediatric neuroendocrine carcinoid tumors: Management, pathology, and imaging findings in a pediatric referral center. Pediatr Blood Cancer 64 (9): , 2017. [PUBMED Abstract]
Pelizzo G, La Riccia A, Bouvier R, et al.: Carcinoid tumors of the appendix in children. Pediatr Surg Int 17 (5-6): 399-402, 2001. [PUBMED Abstract]
Hatzipantelis E, Panagopoulou P, Sidi-Fragandrea V, et al.: Carcinoid tumors of the appendix in children: experience from a tertiary center in northern Greece. J Pediatr Gastroenterol Nutr 51 (5): 622-5, 2010. [PUBMED Abstract]
Henderson L, Fehily C, Folaranmi S, et al.: Management and outcome of neuroendocrine tumours of the appendix-a two centre UK experience. J Pediatr Surg 49 (10): 1513-7, 2014. [PUBMED Abstract]
Dall’Igna P, Ferrari A, Luzzatto C, et al.: Carcinoid tumor of the appendix in childhood: the experience of two Italian institutions. J Pediatr Gastroenterol Nutr 40 (2): 216-9, 2005. [PUBMED Abstract]
Wu H, Chintagumpala M, Hicks J, et al.: Neuroendocrine Tumor of the Appendix in Children. J Pediatr Hematol Oncol 39 (2): 97-102, 2017. [PUBMED Abstract]
Boxberger N, Redlich A, Böger C, et al.: Neuroendocrine tumors of the appendix in children and adolescents. Pediatr Blood Cancer 60 (1): 65-70, 2013. [PUBMED Abstract]
Njere I, Smith LL, Thurairasa D, et al.: Systematic review and meta-analysis of appendiceal carcinoid tumors in children. Pediatr Blood Cancer 65 (8): e27069, 2018. [PUBMED Abstract]
Virgone C, Cecchetto G, Alaggio R, et al.: Appendiceal neuroendocrine tumours in childhood: Italian TREP project. J Pediatr Gastroenterol Nutr 58 (3): 333-8, 2014. [PUBMED Abstract]
de Lambert G, Lardy H, Martelli H, et al.: Surgical Management of Neuroendocrine Tumors of the Appendix in Children and Adolescents: A Retrospective French Multicenter Study of 114 Cases. Pediatr Blood Cancer 63 (4): 598-603, 2016. [PUBMED Abstract]
Extra-appendiceal neuroendocrine (carcinoid) tumors are rare. Most tumors are sporadic, but they may also be part of a hereditary syndrome. A single-institution retrospective review identified 45 cases of neuroendocrine tumors in children and adolescents between 2003 and 2016.[1][Level of evidence C2] In this study, extra-appendiceal primary tumors (n = 9) were associated with a higher risk of metastasis and recurrence. The Tumori Rari in Etá Pediatrica (TREP) group registered 27 patients between 2000 and 2020.[2]
Extra-appendiceal neuroendocrine tumors of the abdomen occur most often in the pancreas but can also occur in the stomach and liver.[2] In the TREP series of 27 cases, 12 occurred in the pancreas and 10 occurred in the bronchi.[2] The most common clinical presentation is an unknown primary site. Extra-appendiceal neuroendocrine tumors are more likely to be larger and higher grade or to present with metastases.[3] Larger tumor size has been associated with a higher risk of recurrence.[1]
The carcinoid syndrome of excessive excretion of somatostatin is characterized by flushing, labile blood pressure, and metastatic spread of the tumor to the liver.[4] Symptoms may be lessened by giving somatostatin analogues, which are available in short-acting and long-acting forms.[5]
Clinical experience with extra-appendiceal neuroendocrine tumors is reported almost entirely in adults. Histopathology is graded by mitotic rate, Ki-67 labeling index, and presence of necrosis into well-differentiated (low grade, G1), moderately differentiated (intermediate grade, G2) and poorly differentiated (high grade, G3) tumors.[6] For more information, see Gastrointestinal Neuroendocrine Tumors Treatment.
Treatment and Outcome of Extra-Appendiceal Gastrointestinal Neuroendocrine Tumors
Complete surgical resection and localized disease are associated with a favorable clinical outcome.[2,7]
In one retrospective single-institution study, the 5-year relapse-free survival rate was 41% for patients with extra-appendiceal neuroendocrine tumors. The overall survival (OS) rate was 66%.[3]
Treatment options for resectable extra-appendiceal neuroendocrine tumors include the following:
SSTR2 ligands include octreotide, long-acting repeatable octreotide, and lanreotide. Octreotide is not practical for therapy because its short half-life necessitates frequent administration. Long-acting, repeatable octreotide and lanreotide have been evaluated in prospective, randomized, placebo-controlled trials.[10,11] Patient age was not specified in the first trial, and eligibility was restricted to age 18 years and older in the second trial. Neither agent produced significant objective responses in measurable tumors. Both agents were associated with statistically significant increases in progression-free survival and time to progression, and both agents are recommended for the treatment of unresectable extra-appendiceal neuroendocrine tumors in adults.
A phase III trial included 231 patients with advanced or metastatic extra-appendiceal neuroendocrine tumors. Patients were randomly assigned to treatment with lutetium Lu 177 (177Lu)-DOTATATE plus long-acting octreotide or high-dose long-acting octreotide (control group). While the median OS did not reach statistical significance, there was an 11.7-month difference, with 48.0 months (95% confidence interval [CI], 37.4–55.2) in the 177Lu-DOTATATE group and 36.3 months (95% CI, 25.9–51.7) in the control group.[16] The U.S. Food and Drug Administration approved the use of 177Lu-DOTATATE for children aged 12 years and older with somatostatin receptor–positive gastroenteropancreatic neuroendocrine tumors.
Embolization, peptide receptor radionuclide therapy, mTOR inhibitors, and TKIs have been used for treatment.[9,12–14]
Conventional cytotoxic chemotherapy appears to be inactive.[3]
References
Degnan AJ, Tocchio S, Kurtom W, et al.: Pediatric neuroendocrine carcinoid tumors: Management, pathology, and imaging findings in a pediatric referral center. Pediatr Blood Cancer 64 (9): , 2017. [PUBMED Abstract]
Virgone C, Ferrari A, Chiaravalli S, et al.: Extra-appendicular neuroendocrine tumors: A report from the TREP project (2000-2020). Pediatr Blood Cancer 68 (4): e28880, 2021. [PUBMED Abstract]
Boston CH, Phan A, Munsell MF, et al.: A Comparison Between Appendiceal and Nonappendiceal Neuroendocrine Tumors in Children and Young Adults: A Single-institution Experience. J Pediatr Hematol Oncol 37 (6): 438-42, 2015. [PUBMED Abstract]
Tormey WP, FitzGerald RJ: The clinical and laboratory correlates of an increased urinary 5-hydroxyindoleacetic acid. Postgrad Med J 71 (839): 542-5, 1995. [PUBMED Abstract]
Delaunoit T, Rubin J, Neczyporenko F, et al.: Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine tumors. Mayo Clin Proc 80 (4): 502-6, 2005. [PUBMED Abstract]
Enzler T, Fojo T: Long-acting somatostatin analogues in the treatment of unresectable/metastatic neuroendocrine tumors. Semin Oncol 44 (2): 141-156, 2017. [PUBMED Abstract]
Courtel T, Orbach D, Lacour B, et al.: Childhood pancreatic neuroendocrine neoplasms: A national experience. Pediatr Blood Cancer 72 (2): e31258, 2025. [PUBMED Abstract]
Ambe CM, Nguyen P, Centeno BA, et al.: Multimodality Management of “Borderline Resectable” Pancreatic Neuroendocrine Tumors: Report of a Single-Institution Experience. Cancer Control 24 (5): 1073274817729076, 2017 Oct-Dec. [PUBMED Abstract]
Elf AK, Andersson M, Henrikson O, et al.: Radioembolization Versus Bland Embolization for Hepatic Metastases from Small Intestinal Neuroendocrine Tumors: Short-Term Results of a Randomized Clinical Trial. World J Surg 42 (2): 506-513, 2018. [PUBMED Abstract]
Rinke A, Wittenberg M, Schade-Brittinger C, et al.: Placebo-Controlled, Double-Blind, Prospective, Randomized Study on the Effect of Octreotide LAR in the Control of Tumor Growth in Patients with Metastatic Neuroendocrine Midgut Tumors (PROMID): Results of Long-Term Survival. Neuroendocrinology 104 (1): 26-32, 2017. [PUBMED Abstract]
Caplin ME, Pavel M, Ćwikła JB, et al.: Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 371 (3): 224-33, 2014. [PUBMED Abstract]
Brabander T, Teunissen JJ, Van Eijck CH, et al.: Peptide receptor radionuclide therapy of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab 30 (1): 103-14, 2016. [PUBMED Abstract]
Gajate P, Martínez-Sáez O, Alonso-Gordoa T, et al.: Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manag Res 9: 215-224, 2017. [PUBMED Abstract]
Liu IH, Kunz PL: Biologics in gastrointestinal and pancreatic neuroendocrine tumors. J Gastrointest Oncol 8 (3): 457-465, 2017. [PUBMED Abstract]
Vellani SD, Nigro A, Varatharajan S, et al.: Emerging Immunotherapeutic and Diagnostic Modalities in Carcinoid Tumors. Molecules 28 (5): , 2023. [PUBMED Abstract]
Strosberg JR, Caplin ME, Kunz PL, et al.: 177Lu-Dotatate plus long-acting octreotide versus high‑dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol 22 (12): 1752-1763, 2021. [PUBMED Abstract]
Metastatic Gastrointestinal Neuroendocrine Tumors
Treatment of metastatic neuroendocrine tumors of the large bowel, pancreas, or stomach is complicated and requires treatment similar to that given for adult high-grade neuroendocrine tumors. For more information about treatment options for patients with malignant carcinoid tumors, see Gastrointestinal Neuroendocrine Tumors Treatment.
Treatment Options Under Clinical Evaluation for Pediatric Gastrointestinal Neuroendocrine Tumors
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has slowly increased since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation to achieve optimal survival and quality of life:
Primary care physicians.
Pediatric surgeons.
Pathologists.
Pediatric radiation oncologists.
Pediatric medical oncologists and hematologists.
Ophthalmologists.
Rehabilitation specialists.
Pediatric oncology nurses.
Social workers.
Child-life professionals.
Psychologists.
Nutritionists.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children and adolescents with cancer.[2] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Other types of clinical trials test novel therapies when there is no standard therapy for a cancer diagnosis. Most of the progress in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[3–5] Childhood and adolescent cancer survivors require close monitoring because side effects of cancer therapy may persist or develop months or years after treatment. For information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
Childhood cancer is a rare disease, with about 15,000 cases diagnosed annually in the United States in individuals younger than 20 years.[6] The U.S. Rare Diseases Act of 2002 defines a rare disease as one that affects populations smaller than 200,000 people in the United States. Therefore, all pediatric cancers are considered rare.
The designation of a rare tumor is not uniform among pediatric and adult groups. In adults, rare cancers are defined as those with an annual incidence of fewer than six cases per 100,000 people. They account for up to 24% of all cancers diagnosed in the European Union and about 20% of all cancers diagnosed in the United States.[7,8] In children and adolescents, the designation of a rare tumor is not uniform among international groups, as follows:
A consensus effort between the European Union Joint Action on Rare Cancers and the European Cooperative Study Group for Rare Pediatric Cancers estimated that 11% of all cancers in patients younger than 20 years could be categorized as very rare. This consensus group defined very rare cancers as those with annual incidences of fewer than two cases per 1 million people. However, three additional histologies (thyroid carcinoma, melanoma, and testicular cancer) with incidences of more than two cases per 1 million people were also included in the very rare group due to a lack of knowledge and expertise in the management of these tumors.[9]
The Children’s Oncology Group defines rare pediatric cancers as those listed in the International Classification of Childhood Cancer subgroup XI, which includes thyroid cancers, melanomas and nonmelanoma skin cancers, and multiple types of carcinomas (e.g., adrenocortical carcinomas, nasopharyngeal carcinomas, and most adult-type carcinomas such as breast cancers and colorectal cancers).[10] These diagnoses account for about 5% of the cancers diagnosed in children aged 0 to 14 years and about 27% of the cancers diagnosed in adolescents aged 15 to 19 years.[4]
Most cancers in subgroup XI are either melanomas or thyroid cancers, with other cancer types accounting for only 2% of the cancers diagnosed in children aged 0 to 14 years and 9.3% of the cancers diagnosed in adolescents aged 15 to 19 years.
These rare cancers are extremely challenging to study because of the relatively few patients with any individual diagnosis, the predominance of rare cancers in the adolescent population, and the small number of clinical trials for adolescents with rare cancers.
Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010. [PUBMED Abstract]
American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed February 25, 2025.
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.
Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr. [PUBMED Abstract]
Gatta G, Capocaccia R, Botta L, et al.: Burden and centralised treatment in Europe of rare tumours: results of RARECAREnet-a population-based study. Lancet Oncol 18 (8): 1022-1039, 2017. [PUBMED Abstract]
DeSantis CE, Kramer JL, Jemal A: The burden of rare cancers in the United States. CA Cancer J Clin 67 (4): 261-272, 2017. [PUBMED Abstract]
Ferrari A, Brecht IB, Gatta G, et al.: Defining and listing very rare cancers of paediatric age: consensus of the Joint Action on Rare Cancers in cooperation with the European Cooperative Study Group for Pediatric Rare Tumors. Eur J Cancer 110: 120-126, 2019. [PUBMED Abstract]
Pappo AS, Krailo M, Chen Z, et al.: Infrequent tumor initiative of the Children’s Oncology Group: initial lessons learned and their impact on future plans. J Clin Oncol 28 (33): 5011-6, 2010. [PUBMED Abstract]
Latest Updates to This Summary (12/26/2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
This summary was reformatted.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of pediatric gastrointestinal neuroendocrine tumors. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Pediatric Gastrointestinal Neuroendocrine Tumors Treatment are:
Denise Adams, MD (Children’s Hospital Boston)
Karen J. Marcus, MD, FACR (Dana-Farber of Boston Children’s Cancer Center and Blood Disorders Harvard Medical School)
William H. Meyer, MD
Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children’s Healthcare of Atlanta – Egleston Campus)
Arthur Kim Ritchey, MD (Children’s Hospital of Pittsburgh of UPMC)
Carlos Rodriguez-Galindo, MD (St. Jude Children’s Research Hospital)
Stephen J. Shochat, MD (St. Jude Children’s Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Leaving ChemoPrescribe
You are now leaving ChemoPrescribe website.
To return to the website, click on the back arrow on your browser.



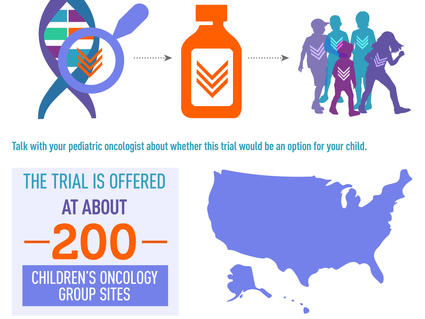

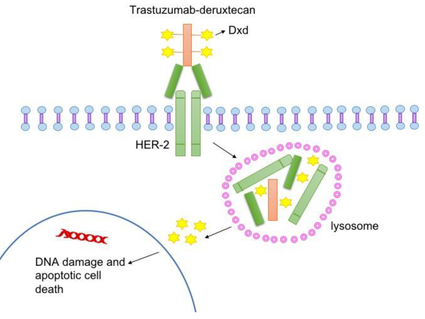
. An inset shows cancer cells spreading from the primary cancer, through the blood and lymph systems, to another part of the body where a metastatic tumor has formed.")


; a brown lesion with a large and irregular border on the skin (panel 2); and a large, asymmetrical, scaly, red and brown lesion on the skin (panel 3).")
. Also shown are the dermis (inner layer of the skin) and the subcutaneous tissue below the dermis.")
 and without ulceration. The panel on the right shows a stage IB tumor that is more than 1 but not more than 2 millimeters thick, without ulceration. Also shown are the epidermis (outer layer of the skin), the dermis (inner layer of the skin), and the subcutaneous tissue below the dermis.")
. The panel on the right shows a tumor that is more than 2 but not more than 4 millimeters thick, without ulceration. Also shown are the epidermis (outer layer of the skin), the dermis (inner layer of the skin), and the subcutaneous tissue below the dermis.")
. There is also an inset that shows 2 millimeters is about the size of a new crayon point and 5 millimeters is about the size of a pencil-top eraser. The panel on the right shows a tumor that is more than 4 millimeters thick, without ulceration. There is also an inset that shows 5 millimeters is about the size of a pencil-top eraser. Also shown are the epidermis (outer layer of the skin), the dermis (inner layer of the skin), and the subcutaneous tissue below the dermis.")
. Also shown are the epidermis (outer layer of the skin), the dermis (inner layer of the skin), and the subcutaneous tissue below the dermis.")
 tract, bone, muscle, and distant lymph nodes. An inset shows cancer cells spreading through the blood and lymph system to another part of the body where a metastatic tumor has formed.")

; the third panel shows the removal of the tumor and the sentinel nodes to check for cancer cells.")
 compared to the size of a pea (1 cm), a peanut (2 cm), a grape (3 cm), a walnut (4 cm), a lime (5 cm), an egg (6 cm), a peach (7 cm), and a grapefruit (10 cm). Also shown is a 10-cm ruler and a 4-inch ruler.")



