During the past five decades, dramatic progress has been made in the development of curative therapies for pediatric malignancies. More than 80% of children with cancer who have access to contemporary therapies are expected to survive into adulthood.[1] The therapies responsible for this survival can also produce adverse, long-term, health-related outcomes, referred to as late effects, which appear months to years after completion of cancer treatment.
Many approaches have been used to study the very long-term morbidity associated with childhood cancer and its contribution to early mortality. These initiatives have used a spectrum of resources, including data from the following:
High-quality data is needed to establish the occurrence of and risk profiles for late cancer treatment–related toxicity. The highest quality data typically comes from studies that report outcomes in survivors who have undergone medical assessments that provide well-characterized clinical statuses, treatment exposures, and specific late effects. Regardless of study methodology, it is important to consider selection and participation bias of the cohort studies in the context of the findings.
Prevalence of Late Effects in Childhood Cancer Survivors
Late effects are common in adults who have survived childhood cancer. Their prevalence increases as time from cancer diagnosis elapses. Multi-institutional and population-based studies have shown excess risk of hospital-related morbidity among childhood and young adult cancer survivors compared with age- and sex-matched controls, with some evidence that this risk is disproportionately high among survivors of racial and ethnic minority populations.[3,9–13]
Among adults who were treated for cancer during childhood, late effects contribute to a high burden of morbidity. Research has shown the following:[4,7,14–18]
60% to more than 90% of survivors develop one or more chronic health conditions.
20% to 80% of survivors experience severe or life-threatening complications during adulthood.
Morbidity accumulation is accelerated in young adult survivors of childhood cancer, compared with that of siblings and the general population. Accumulation of chronic diseases predicts risk of early mortality.[19]
The St. Jude Life (SJLIFE) cohort study aimed to describe the cumulative burden of cancer therapy using the cumulative burden metric, which incorporates multiple health conditions and recurrent events into a single metric that takes into account competing risks. By age 50 years, survivors in this cohort experienced an average of 17.1 chronic health conditions, 4.7 of which were severe/disabling, life threatening, or fatal.[16] This finding contrasts with the cumulative burden in matched community controls, who experienced 9.2 chronic health conditions, 2.3 of which were severe/disabling, life threatening, or fatal (see Figure 1).[16]
EnlargeFigure 1. Figure shows distribution of cumulative burden by age among childhood cancer survivors of specific pediatric cancer subtypes and community controls participating in the SJLIFE cohort study. The cumulative burden at age 30 years and rate of cumulative burden growth is variable across cancer subtypes and organ systems. Reprinted from The Lancet, Volume 390, Issue 10112, Bhakta N, Liu Q, Ness KK, Baassiri M, Eissa H, Yeo F, Chemaitilly W, Ehrhardt MJ, Bass J, Bishop MW, Shelton K, Lu L, Huang S, Li Z, Caron E, Lanctot J, Howell C, Folse T, Joshi V, Green DM, Mulrooney DA, Armstrong GT, Krull KR, Brinkman TM, Khan RB, Srivastava DK, Hudson MM, Yasui Y, Robison LL, The cumulative burden of surviving childhood cancer: an initial report from the St Jude Lifetime Cohort Study (SJLIFE), Pages 2569–2582, Copyright (2017), with permission from Elsevier.
SJLIFE cohort study investigators compared the cumulative burden of chronic health conditions among 4,612 adolescent and young adult survivors at the ages of 18 years (the time of transition from pediatric to adult health care systems) and 26 years (the time of transition from family to individual health insurance plans) with that of 625 controls.[20]
Survivors at the age of 18 years experienced an average of 22.3 disabling conditions per 100 individuals versus 3.5 in controls, and 128.7 lower-severity conditions (at risk of progressing to higher-grade disabling conditions) versus 12.4 in controls.
Survivors at the age of 26 years experienced an average of 40.3 disabling conditions per 100 individuals versus 5.7 in controls, and 240.5 lower-severity conditions versus 51.3 in controls.
The cumulative burden of disabling, disease-specific conditions at the ages of 18 and 26 years was most notable for survivors of bone tumors (musculoskeletal: 99.9 and 121.70, respectively), soft tissue sarcomas (musculoskeletal: 49.5 and 54.1, respectively), and central nervous system (CNS) tumors (neurological: 24.7 and 36.8, respectively).
The cumulative burden of lower-severity conditions (potentially amenable to intervention) at the ages of 18 and 26 years was most notable for neurological conditions across most cancer subgroups, with the highest cumulative burden in CNS tumor survivors (95.2 and 162.3, respectively).
These findings highlight the importance of optimizing access to health care and health insurance as survivors age and can no longer participate in pediatric health care systems.
The variability in prevalence is related to differences in the following:
Age and follow-up time of the cohorts studied.
Methods and consistency of assessment (e.g., self-reported vs. risk-based medical evaluations).
Treatment intensity and treatment era.
Childhood Cancer Survivor Study (CCSS) investigators demonstrated that the elevated risk of morbidity and mortality among aging survivors in the cohort increases beyond the fourth decade of life. By age 50 years, the cumulative incidence of a self-reported severe, disabling, life-threatening, or fatal health condition was 53.6% among survivors, compared with 19.8% among a sibling control group. Among survivors who reached age 35 years without a previous severe, disabling, life-threatening, or fatal health condition, 25.9% experienced a new grade 3 to grade 5 health condition within 10 years, compared with 6.0% of healthy siblings.[4]
The presence of serious, disabling, and life-threatening chronic health conditions adversely affects the health status of aging survivors. The greatest impact is on functional impairment and activity limitations. Predictably, chronic health conditions have been reported to contribute to a higher prevalence of emotional distress symptoms in adult survivors than in population controls.[21] Female survivors demonstrate a steeper trajectory of age-dependent decline in health status than do male survivors.[22] The even-higher prevalence of late effects among cohorts evaluated by clinical assessments is related to the subclinical and undiagnosed conditions detected by screening and surveillance measures.[7]
CCSS investigators also evaluated the impact of race and ethnicity on late outcomes. The study compared late mortality, subsequent neoplasms, and chronic health conditions in Hispanic (n = 750) and non-Hispanic Black (n = 694) participants with those in non-Hispanic White participants (n = 12,397).[23] The following results were observed:
Cancer treatment did not account for disparities in mortality, chronic health conditions, or subsequent neoplasms observed among the groups.
Differences in socioeconomic status and cardiovascular risk factors affected risk. All-cause mortality was higher among non-Hispanic Black participants than among other groups, but this difference disappeared after adjustment for socioeconomic status.
Risk of developing diabetes was elevated among racial and ethnic minority groups even after adjustment for socioeconomic and obesity status.
Non-Hispanic Black patients had a higher likelihood of reporting cardiac conditions, but this risk diminished after adjusting for cardiovascular risk factors.
Nonmelanoma skin cancer was not reported by non-Hispanic Black participants, a finding that has been replicated by other studies.[24] Hispanic participants had a lower risk of nonmelanoma skin cancer than non-Hispanic White participants.
Recognition of late effects, concurrent with advances in cancer biology, radiological sciences, and supportive care, has resulted in a change in the prevalence and spectrum of treatment effects. In an effort to reduce and prevent late effects, contemporary therapy for most pediatric malignancies has evolved to a risk-adapted approach that is assigned on the basis of a variety of clinical, biological, and sometimes genetic factors.
The CCSS reported that with decreased cumulative dose and frequency of therapeutic radiation from 1970 to 1999, survivors have experienced a significant decrease in risk of subsequent neoplasms.[25]
With the exception of survivors requiring intensive multimodality therapy for refractory/relapsed malignancies, life-threatening treatment effects are relatively uncommon after contemporary therapy in early follow-up (up to 10 years after diagnosis).
However, survivors still frequently experience life-altering morbidity related to effects of cancer treatment on endocrine, reproductive, musculoskeletal, and neurological function.
A CCSS investigation examined temporal patterns in the cumulative incidence of severe to fatal chronic health conditions among survivors treated from 1970 to 1999.[26]
The 20-year cumulative incidence of at least one grade 3 to 5 chronic condition decreased significantly, from 33.2% for survivors diagnosed between 1970 and 1979, to 29.3% for those diagnosed between 1980 and 1989, to 27.5% for those diagnosed between 1990 and 1999, compared with a 4.6% incidence in a sibling cohort.
The overall decrease in incidence of chronic conditions across the three treatment decades was, in part, because of a substantial reduction of endocrinopathies, subsequent malignant neoplasms (SMNs), musculoskeletal conditions, and gastrointestinal conditions, whereas the cumulative incidence of hearing loss increased during this time.
Declines in morbidity were not uniform across the diagnosis groups or condition types because of differences in treatment and survival patterns over time. For more information, see Figure 2.
Despite declines in chronic health conditions over time, self-reported health status has not improved in more recent treatment eras. This finding may be because of the survival of children with higher-risk disease who would have previously died of cancer, or an enhanced awareness of and surveillance for late effects among more recently treated survivors.[27]
EnlargeFigure 2. Cumulative incidence of grade 3–5 chronic health conditions in 5-year survivors of childhood cancer by diagnosis decade and siblings. (A) Cumulative incidence of a first grade 3–5 condition. (B) Cumulative incidence of two or more grade 3–5 conditions. The shaded area represents the 95% confidence interval (CI). The number of participants at risk (number censored) at each 5-year interval post-diagnosis is listed below the x-axis. The number censored does not include those who experienced a competing risk event (death from a cause other than a grade 5 chronic condition). Reprinted from The Lancet Oncology, Volume 19, Issue 12, Todd M Gibson, Sogol Mostoufi-Moab, Kayla L Stratton, Wendy M Leisenring, Dana Barnea, Eric J Chow, Sarah S Donaldson, Rebecca M Howell, Melissa M Hudson, Anita Mahajan, Paul C Nathan, Kirsten K Ness, Charles A Sklar, Emily S Tonorezos, Christopher B Weldon, Elizabeth M Wells, Yutaka Yasui, Gregory T Armstrong, Leslie L Robinson, Kevin C Oeffinger, Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970–99: a report from the Childhood Cancer Survivor Study cohort. Pages 1590-1601, Copyright (2018), with permission from Elsevier.
Mortality
Late effects also contribute to an excess risk of premature death among long-term survivors of childhood cancer, as observed in the following studies:
Several studies of large cohorts of survivors have reported early mortality among individuals treated for childhood cancer, compared with age- and sex-matched general population controls. Relapsed/refractory primary cancer remains the most frequent cause of death, followed by excess cause-specific mortality from subsequent primary cancers, and cardiac and pulmonary toxicity.[28–33]
A CCSS study evaluated specific health-related causes of late mortality and excess deaths, compared with the general U.S. population to identify targets to reduce future risk.[33]
At a median follow-up of 29 years from diagnosis, the 40-year cumulative all-cause mortality rate was 23.3%, with 51.2% of deaths attributed to health-related causes.
Survivors who were 40 or more years from diagnosis experienced 131 excess health-related deaths per 10,000 person-years (95% confidence interval [CI], 111–163). Excess deaths were most commonly related to cancer (absolute excess risk per 10,000 person-years, 54; 95% CI, 41–68), heart disease (27; 18–38), and cerebrovascular disease (10; 5–17).
Healthy lifestyle (assessed by smoking status, alcohol consumption, physical activity, body mass index, and absence of hypertension and diabetes) were each associated with a 20% to 30% reduction in health-related mortality, independent of other factors.
An analysis of the CCSS and Surveillance, Epidemiology, and End Results (SEER) Program data evaluated conditional survival. The study demonstrated a subsequent 5-year survival rate of 92% or higher among most diagnoses at 5 years, 10 years, 15 years, and 20 years. Among those who had survived at least 5 years from diagnosis, the probability of all-cause mortality in the next 10 years was 8.8% in the CCSS and 10.6% in the SEER study, with neoplasms accounting for cause of death in approximately 75% of survivors.[34]
Despite high premature morbidity rates, overall mortality has decreased over time.[28,30,31,35,36]
CCSS investigators evaluated all-cause and health-related late mortality (including late effects of cancer therapy), SMNs, chronic health conditions, and neurocognitive outcomes among 6,148 survivors of childhood acute lymphoblastic leukemia (median age, 27.9 years; range, 5.9–61.9 years) diagnosed between 1970 and 1999.[37]
Overall, the 20-year all-cause late mortality rate was 6.6%.
Compared with participants who were treated in the 1970s, patients who were treated with risk-stratified regimens in the 1990s experienced lower health-related late mortality (rate ratio: 1990s standard risk, 0.2; 1990s high risk, 0.3), which was comparable to the U.S. population (standardized mortality ratio [SMR]: 1990s standard risk, 1.3; 1990s high risk, 1.7).
The risk of late mortality and serious chronic health conditions have decreased over time among survivors of acute myeloid leukemia (AML). CCSS investigators evaluated the long-term morbidity, mortality, and health status of more than 800 5-year survivors of childhood AML based on treatment and treatment era. Survivors were compared by treatment group (hematopoietic stem cell transplant [HSCT]); chemotherapy with cranial radiation [CRT]; chemotherapy only) and decade of diagnosis.[38]
Among 856 survivors, the 20-year late mortality cumulative incidence was highest after HSCT (13.9%; chemotherapy with CRT, 7.6%; chemotherapy only, 5.1%).
Mortality cumulative incidence decreased for survivors of HSCT diagnosed in the 1990s (8.5%) compared with the 1970s (38.9%), as did standardized mortality rates.
Self-reported health status was good to excellent for 88.2% of childhood AML survivors, regardless of treatment.
Most survivors did not experience any grade 3 to 5 chronic health conditions after 20 years (HSCT, 45.8%; chemotherapy with CRT, 23.7%; chemotherapy only, 27.0%).
A temporal reduction in chronic health conditions cumulative incidence was seen after HSCT (1970s, 76.1%; 1990s, 38.3%; P = .02), mirroring a reduction in total-body irradiation use.
Population-based data from a state cancer registry was used to evaluate differences in survival and long-term outcomes by race and ethnicity among 4,222 children diagnosed with cancer between 1987 and 2012.[12]
Compared with non-Hispanic White children, hospitalization was 70% (hazard ratio [HR], 1.7) more common 5 or more years after diagnosis for American Indian and Alaskan Native children and 50% (HR, 1.5) more common for Black children.
Among survivors at 5 or more years from diagnosis, 2.3-fold to 3.6-fold statistically significant relative increases were observed for hospitalizations for specific conditions for American Indian and Alaskan Native children (HR, 2.3 for infection-related conditions; HR, 3.0 for hematologic-related conditions; HR, 2.6 for digestive-related conditions). The greatest increases were noted for mental health–related conditions (HR, 3.6), a pattern also noted for Black children (HR, 2.5).
An SJLIFE cohort study explored associations between modifiable chronic health conditions and late mortality within the context of social determinants of health.[39]
Among 9,440 5-year childhood cancer survivors included in the analysis (median age at last follow-up, 27.5 years; median follow-up, 18.8 years), all-cause mortality (SMR, 7.6; 95% CI, 7.2–8.1) and health-related late mortality (SMR, 7.6; 95% CI, 7.0–8.2) was significantly higher than expected for U.S. mortality rates.
Among 3,407 adult participants who completed an on-campus assessment (median age at assessment, 35.4 years; median follow-up, 27.3 years), significant increases in late all-cause and health-related deaths were associated with the number of modifiable chronic health conditions, living in a U.S. census block associated with a high area deprivation index, and frailty.
Specific associations for excess health-related mortality included: one grade 2 modifiable chronic health condition (rate ratio [RR], 2.2; 95% CI, 1.1–4.4), two grade 2 modifiable chronic health conditions (RR, 2.5; 95% CI, 1.2–5.2), three grade 2 modifiable chronic health conditions (RR, 4.0; 95% CI, 1.9–8.4), area deprivation index in 51st to 80th percentile (RR, 9.2; 95% CI, 1.2–69.7), area deprivation index in 81st to 100th percentile (RR, 16.2; 95% CI, 2.1–123.7), and frailty (RR, 2.3; 95% CI, 1.2–4.1).
The CCSS and an SJLIFE cohort study investigated the contribution of cancer-predisposing variants to the risk of SMN-related late mortality (5 years or more after diagnosis).[40]
Among 12,469 participants (6,172 male and 6,297 female), including 4,402 from the SJLIFE cohort (median follow-up time since collection of first biospecimen, 7.4 years) and 8,067 from the CCSS cohort (median follow-up time since collection of the first biospecimen, 12.6 years), 641 (5.1%) carried cancer-predisposing variants.
Cancer-predisposing variants were significantly associated with an increased severity of SMNs (common terminology criteria for adverse events grade ≥4 vs. grade <4: odds ratio [OR], 2.15; 95% CI, 1.18–4.19).
SMN-related deaths occurred in 263 participants (2.1%), and other-cause deaths occurred in 426 survivors (3.4%).
At 10 years after the first biospecimen collection, the cumulative SMN-related mortality rate in carriers of cancer-predisposing variants was 3.7% (95% CI, 1.2%–8.5%) in SJLIFE and 6.9% (4.1%–10.7%) in CCSS. In comparison, the cumulative SMN-related mortality rate in noncarriers was 1.5% (1.0%–2.1%) in SJLIFE and 2.1% (1.7%–2.5%) in CCSS.
Carrying a cancer-predisposing variant was associated with an increased risk of SMN-related mortality (SJLIFE: HR, 3.40; 95% CI, 1.37–8.43; CCSS: HR, 3.58; 95% CI, 2.27–5.63).
Survivors of adolescent and young adult (AYA) cancers
Little information is available on late mortality among survivors of AYA cancer.[41–44]
Using SEER data, conditional relative survival up to 25 years after diagnosis was studied in a cohort of AYA patients (N = 205,954) diagnosed with a first malignant cancer (thyroid, melanoma, testicular, breast, lymphoma, leukemia, and CNS tumors).[41]
For all cancer types combined, among individuals who survived up to 5 years, the subsequent 5-year relative survival rate exceeded 95% by 7 years after diagnosis.
Most AYA cancer patients who survived at least 7 years after diagnosis experienced little difference in survival compared with the general population.
For specific cancer types, including CNS tumors, female breast cancer, Hodgkin lymphoma, and leukemia, evidence of excess mortality risk persisted, or re-emerged, more than 10 years after a cancer diagnosis.
Conditional relative survival was lowest for AYA patients with CNS tumors, although patients aged 15 to 29 years demonstrated a higher survival rate than did patients aged 30 to 39 years at the time of diagnosis of their CNS tumors.
A separate analysis of 5-year survivors of AYA cancer (aged 15–39 years at diagnosis), also using SEER data (N = 282,969), demonstrated the following:[42]
The 10-year all-cause mortality rate decreased from 8.3% for those diagnosed between 1975 and 1984 to 5.4% for those diagnosed between 2005 and 2011.
The decrease in mortality primarily resulted from fewer deaths from the initial cancer.
CCSS investigators compared chronic health conditions and all-cause and cause-specific mortality among 5,804 survivors of early-AYA cancer survivors (cancer diagnosis, age 15–20 years; median age, 42 years) and 5,804 childhood cancer survivors (cancer diagnosis, age <15 years; median age, 34 years) matched on primary cancer diagnosis.[43]
The SMR was 5.9 (95% CI, 5.5–6.2) for early-AYA survivors and 6.2 (95% CI, 5.8–6.6) for younger childhood cancer survivors, compared with the general population.
Early-AYA survivors had lower SMRs for death from health-related causes than did childhood cancer survivors (SMR, 4.8 [95% CI, 4.4–5.1] vs. 6.8 [95% CI, 6.2–7.4]), which was primarily evident more than 20 years after cancer diagnosis.
Early-AYA and childhood cancer survivors were at greater risk of developing severe and disabling, life-threatening, or fatal (grades 3–5) health conditions than were siblings of the same age (HR, 4.2 [95% CI, 3.7–4.8] for early-AYA and 5.6 [95% CI, 4.9–6.3] for childhood cancer survivors), although the risk was lower for early-AYA survivors than for childhood cancer survivors.
In a retrospective, population-based cohort study from Kaiser Permanente, cause-specific mortality in 2-year survivors (N = 10,574) of AYA cancers (patients aged 13–39 years who were diagnosed between 1990 and 2012) was examined and compared with individuals without cancer.[45]
AYA cancer survivors were at a 10.4-fold increased risk of death compared with the matched noncancer cohort, and this risk remained elevated at more than 20 years after diagnosis (incidence rate ratio [IRR], 2.9).
Beginning at 15 years after diagnosis, the incidence of second cancer–related mortality exceeded the rate of recurrence-related mortality.
Mortality risk of suicide was doubled in AYA cancer survivors compared with the noncancer cohort.
Chronic comorbidities were investigated in a retrospective, population-based cohort study of 6,778 2-year AYA cancer survivors diagnosed and monitored at Kaiser Permanente.[46]
Approximately 17% of the survivors developed more than one comorbidity. The most common comorbidities were dyslipidemia (22 per 1,000 person-years), hypertension (16 per 1,000 person-years), diabetes (10 per 1,000 person-years), thyroid disorders (9 per 1,000 person-years), and severe depression or anxiety (8 per 1,000 person-years).
IRRs were higher in survivors than in controls without a history of cancer for avascular necrosis (IRR, 8.25), followed by osteoporosis (IRR, 5.75), joint replacement (IRR, 3.89), stroke (IRR, 3.19), premature ovarian failure (IRR, 2.87), and cardiomyopathy or heart failure (IRR, 2.64).
For survivors of AYA cancer, the prevalence of multiple comorbidities approached 40% at 10 years after index date (a 2-year time point from diagnosis), compared with 20% for those without cancer (P < .001).
Monitoring for Late Effects
Recognition of both acute and late modality–specific toxicity has motivated investigations evaluating the pathophysiology and prognostic factors for cancer treatment–related effects. Consequently, the results of late effects research have played an important role in the following areas:
Changing pediatric cancer therapeutic approaches to reduce treatment-related mortality among survivors treated in more recent eras.[47]
The development of risk counseling and health screening recommendations for long-term survivors by identifying the clinical and treatment characteristics of those at highest risk of therapy-related complications.[48]
The common late effects of pediatric cancer encompass several broad domains, including the following:
Growth and development.
Organ function.
Reproductive capacity and health of offspring.
Secondary carcinogenesis.
Psychosocial sequelae related to the primary cancer, its treatment, or maladjustment associated with the cancer experience.
Late sequelae of therapy for childhood cancer can be anticipated based on therapeutic exposures, but the magnitude of risk and the manifestations in an individual patient are influenced by numerous factors. Multiple factors should be considered in the risk assessment for a given late effect (see Figure 3).[49]
Cancer-related factors:
Organs or tissues affected by the cancer.
Direct tissue effects.
Cancer-induced organ dysfunction or other tissue effects.
Treatment-related factors:
Radiation therapy: Total dose, fraction size, organ or tissue volume exposed.
The need for long-term follow-up of childhood cancer survivors is supported by the American Society of Pediatric Hematology/Oncology, the International Society of Pediatric Oncology, the American Academy of Pediatrics, the Children’s Oncology Group (COG), and the Institute of Medicine. A risk-based medical follow-up is recommended, which includes a systematic plan for lifelong screening, surveillance, and prevention that incorporates risk estimates based on the following:[49]
Previous cancer.
Cancer therapy.
Genetic predisposition.
Lifestyle behaviors.
Comorbid conditions.
Sex.
Part of long-term follow-up also focuses on appropriate screening of educational and vocational progress. Specific treatments for childhood cancer, especially those that directly impact nervous system structures, may result in sensory, motor, and neurocognitive deficits that may have adverse effects on functional status, educational attainment, and future vocational opportunities. In support of this, a CCSS investigation observed the following:[50]
Treatment with cranial radiation doses of 25 Gy or higher was associated with higher odds of unemployment (health related: OR, 3.47; 95% CI, 2.54–4.74; seeking work: OR, 1.77; 95% CI, 1.15–2.71).
Unemployed survivors reported higher levels of poor physical functioning than employed survivors, had lower education and income, and were more likely to be publicly insured than unemployed siblings.
These data emphasize the importance of facilitating survivor access to individualized education services, which has been demonstrated to have a positive impact on education achievement.[51] These services may in turn enhance vocational opportunities.
In addition to risk-based screening for medical late effects, the impact of health behaviors on cancer-related health risks is also emphasized. Health-promoting behaviors are stressed for survivors of childhood cancer. Educational efforts focused on healthy lifestyle behaviors include the following:
Abstinence from smoking, excess alcohol use, and illicit drug use to reduce the risk of organ toxicity and, potentially, subsequent neoplasms.
Healthy dietary practices (e.g., a diet rich in plant foods and moderate in animal foods) [52] and active lifestyle to reduce treatment-related metabolic and cardiovascular complications.
Regular physical activity to reduce neurocognitive problems and enhance psychological outcomes.[53,54]
Survivors who engaged in consistent physical activity over time had fewer neurocognitive problems, including difficulties with task efficiency, emotional regulation, organization, and memory. They also experienced larger neurocognitive improvements, compared with those who had inconsistent activity levels.[53]
Vigorous exercise in survivors has been associated with a lower prevalence of depression and somatization, as well as less impairment in physical functioning, general health and vitality, emotional role limitations, and mental health quality-of-life domains.[54]
Proactively addressing unhealthy and risky behaviors is pertinent because several research investigations confirm that long-term survivors use tobacco and alcohol and have inactive lifestyles despite their increased risk of cardiac, pulmonary, and metabolic late effects.[55–57]
Access to risk-based survivor care
Most childhood cancer survivors do not receive recommended risk-based care. The CCSS observed the following:
92.8% of survivors reported receiving some form of medical care in the previous year.[58]
Nearly 40% reported receiving care that focused on their previous cancer (survivor-focused care).[58]
Surveillance for new cases of cancer was very low in survivors at the highest risk of colon, breast, or skin cancer, suggesting that survivors and their physicians need education about the risk of subsequent neoplasms and recommended surveillance.[59]
Sociodemographic factors have been linked to declining rates of follow-up care over time from diagnosis. CCSS participants who were male, had a household income of less than $20,000 per year, and had lower educational attainment (high school education or less) were more likely to report no care at their most recent follow-up survey. This trend is concerning because the prevalence of chronic health conditions increases with longer elapsed time from cancer diagnosis in adults treated for cancer during childhood.[60]
A study that included 975 adult survivors of childhood cancer identified factors associated with attending the recommended risk-based, cancer-related medical visits. The relative risk of having a cancer-related visit was higher among survivors who:[61]
Assigned a greater importance to these visits.
Perceived a greater susceptibility to health problems.
Had experienced a cancer-related chronic health problem that was moderate to life-threatening.
Were seeing a primary care provider for a cancer-related problem.
Had received a cancer treatment care plan.
Expressed greater confidence in physicians’ abilities to address questions and concerns.
Access to health insurance appears to play an important role in risk-based survivor care.[62,63] Lack of access to health insurance affects the following:
Cancer-related visits. In the CCSS, uninsured survivors were less likely than those privately insured to report a cancer-related visit (adjusted relative risk, 0.83; 95% CI, 0.75–0.91) or a cancer center visit (adjusted relative risk, 0.83; 95% CI, 0.71–0.98). Uninsured survivors had lower levels of utilization in all measures of care than privately insured survivors. In contrast, publicly insured survivors were more likely to report a cancer-related visit (adjusted relative risk, 1.22; 95% CI, 1.11–1.35) or a cancer center visit (adjusted relative risk, 1.41; 95% CI, 1.18–1.70) than were privately insured survivors.[62]
Health care outcomes. In a study comparing health care outcomes for long-term AYA cancer survivors with young adults who have no cancer history, the proportion of uninsured survivors did not differ between the two groups.[64]
Financial burden. Subgroups of adult survivors of childhood cancer may be at additional risk of health care barriers due to financial hardship. Younger survivors (aged 20–29 years), females, non-White survivors, and survivors reporting poorer health faced more cost barriers, which may inhibit the early detection of late effects.[64] Survivors are more likely than their siblings to forego needed medical care related to financial challenges.[65]
Overall, lack of health insurance—related to health issues, unemployment, and other societal factors—remains a significant concern for survivors of childhood cancer.[66,67] Legislation, including the Health Insurance Portability and Accountability Act (HIPAA),[68,69] has improved access and retention of health insurance among survivors, although the quality and limitations associated with these policies have not been well studied.
Transition to Survivor Care
Long-term follow-up programs
Transition of care from the pediatric to adult health care setting is necessary for most childhood cancer survivors in the United States.
When available, multidisciplinary long-term follow-up programs in the pediatric cancer center work collaboratively with community physicians to provide care for childhood cancer survivors. This type of shared care has been proposed as the optimal model to facilitate coordination between the cancer center oncology team and community physician groups providing survivor care.[70]
An essential service of long-term follow-up programs is the organization of an individualized survivorship care plan that includes the following:
Details about therapeutic interventions undertaken for childhood cancer and their potential health risks (e.g., chemotherapy type and cumulative dose, radiation treatment fields and dose, surgical procedures, blood product transfusions, and HSCT).
Personalized health screening recommendations.
Information about lifestyle factors that modify risks.
A CCSS investigation that evaluated perceptions of future health and cancer risk highlighted the importance of continuing education of survivors during long-term follow-up evaluations. A substantial subgroup of adult survivors reported a lack of concern about future health (24%) and subsequent cancer risks (35%), even after exposure to treatments associated with increased risks. These findings present concerns that survivors may be less likely to engage in beneficial screenings and risk-reduction activities.[71]
The CCSS evaluated the surveillance and screening practices of 11,337 childhood cancer survivors. They found that fewer than half of high-risk survivors at increased risk of developing SMNs or cardiac dysfunction received the recommended surveillance, which likely exposes them to preventable morbidity and mortality.[59]
27% of survivors and 20% of primary care providers (PCP) had a survivorship care plan. Survivors treated after 1990 were more likely to have a survivorship care plan.
Survivorship care plan possession by high-risk survivors was associated with increased adherence to COG-recommended breast (22% vs. 8%), skin (35% vs. 23%), and cardiac (67% vs. 33%) surveillance. PCP survivorship care plan possession was associated with increased adherence to skin surveillance (40% vs. 23%).
Among high-risk survivors, adherence increased for colorectal (14% to 41%, P < .001) and cardiac (22% to 38%, P < .001) surveillance and decreased for breast surveillance (38% to 13%, P < .001) between 2007 and 2016.
For average-risk survivors, better adherence to American Cancer Society recommendations for breast (57%), cervical (84%), and colorectal (69%) screening was observed than with COG recommendations. PCP survivorship care plan possession was associated with increased adherence to breast and colorectal screening. Survivors were less adherent to breast screening than the general population and less adherent to cervical screening than siblings.
For survivors who have not been provided with this information, the COG offers a template that can be used by survivors to organize a personal treatment summary. For more information, see the COG Survivorship Guidelines, Appendix 1.
COG long-term follow-up guidelines for childhood and AYA cancer survivors
To facilitate survivor and provider access to succinct information to guide risk-based care, COG investigators have organized a compendium of exposure- and risk-based health surveillance recommendations, with the goal of standardizing the care of childhood cancer survivors.[72]
The compendium of resources includes the following:
Health Links. Patient education materials called Health Links provide detailed information on guideline-specific topics to enhance health maintenance and promotion among this population of cancer survivors.[73]
Information concerning late effects is summarized in tables throughout this summary.
Several groups have undertaken research to evaluate the yield from risk-based screening as recommended by the COG and other pediatric oncology cooperative groups.[7,74,75] Pertinent considerations in interpreting the results of these studies include the following:
Variability in the cohort’s age at treatment.
Age at screening.
Time from cancer treatment.
Participation bias.
Collectively, these studies demonstrate that screening identifies a substantial proportion of individuals with previously unrecognized, treatment-related health complications of varying degrees of severity. Study results have also identified low-yield evaluations that have encouraged revisions of screening recommendations. Ongoing research is evaluating the cost effectiveness of screening in the context of consideration of benefits, risks, and harms.
References
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
Lorenzi MF, Xie L, Rogers PC, et al.: Hospital-related morbidity among childhood cancer survivors in British Columbia, Canada: report of the childhood, adolescent, young adult cancer survivors (CAYACS) program. Int J Cancer 128 (7): 1624-31, 2011. [PUBMED Abstract]
Rebholz CE, Reulen RC, Toogood AA, et al.: Health care use of long-term survivors of childhood cancer: the British Childhood Cancer Survivor Study. J Clin Oncol 29 (31): 4181-8, 2011. [PUBMED Abstract]
Armstrong GT, Kawashima T, Leisenring W, et al.: Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol 32 (12): 1218-27, 2014. [PUBMED Abstract]
Teepen JC, Kok JL, Feijen EAM, et al.: Questionnaire- and linkage-based outcomes in Dutch childhood cancer survivors: Methodology of the DCCSS LATER study part 1. Cancer Med 12 (6): 7588-7602, 2023. [PUBMED Abstract]
Geenen MM, Cardous-Ubbink MC, Kremer LC, et al.: Medical assessment of adverse health outcomes in long-term survivors of childhood cancer. JAMA 297 (24): 2705-15, 2007. [PUBMED Abstract]
Hudson MM, Ness KK, Gurney JG, et al.: Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA 309 (22): 2371-81, 2013. [PUBMED Abstract]
Feijen EAM, Teepen JC, van Dulmen-den Broeder E, et al.: Clinical evaluation of late outcomes in Dutch childhood cancer survivors: Methodology of the DCCSS LATER 2 study. Pediatr Blood Cancer 70 (5): e30212, 2023. [PUBMED Abstract]
Kurt BA, Nolan VG, Ness KK, et al.: Hospitalization rates among survivors of childhood cancer in the Childhood Cancer Survivor Study cohort. Pediatr Blood Cancer 59 (1): 126-32, 2012. [PUBMED Abstract]
Zhang Y, Lorenzi MF, Goddard K, et al.: Late morbidity leading to hospitalization among 5-year survivors of young adult cancer: a report of the childhood, adolescent and young adult cancer survivors research program. Int J Cancer 134 (5): 1174-82, 2014. [PUBMED Abstract]
Sørensen GV, Winther JF, de Fine Licht S, et al.: Long-Term Risk of Hospitalization Among Five-Year Survivors of Childhood Leukemia in the Nordic Countries. J Natl Cancer Inst 111 (9): 943-951, 2019. [PUBMED Abstract]
Emerson MA, Olshan AF, Chow EJ, et al.: Hospitalization and Mortality Outcomes Among Childhood Cancer Survivors by Race, Ethnicity, and Time Since Diagnosis. JAMA Netw Open 5 (6): e2219122, 2022. [PUBMED Abstract]
Streefkerk N, Tissing WJE, Korevaar JC, et al.: A detailed insight in the high risks of hospitalizations in long-term childhood cancer survivors-A Dutch LATER linkage study. PLoS One 15 (5): e0232708, 2020. [PUBMED Abstract]
Berbis J, Michel G, Chastagner P, et al.: A French cohort of childhood leukemia survivors: impact of hematopoietic stem cell transplantation on health status and quality of life. Biol Blood Marrow Transplant 19 (7): 1065-72, 2013. [PUBMED Abstract]
Phillips SM, Padgett LS, Leisenring WM, et al.: Survivors of childhood cancer in the United States: prevalence and burden of morbidity. Cancer Epidemiol Biomarkers Prev 24 (4): 653-63, 2015. [PUBMED Abstract]
Bhakta N, Liu Q, Ness KK, et al.: The cumulative burden of surviving childhood cancer: an initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet 390 (10112): 2569-2582, 2017. [PUBMED Abstract]
Salloum R, Chen Y, Yasui Y, et al.: Late Morbidity and Mortality Among Medulloblastoma Survivors Diagnosed Across Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 37 (9): 731-740, 2019. [PUBMED Abstract]
Streefkerk N, Teepen JC, Feijen EAM, et al.: The cumulative burden of self-reported, clinically relevant outcomes in long-term childhood cancer survivors and implications for survivorship care: A DCCSS LATER study. Cancer 130 (8): 1349-1358, 2024. [PUBMED Abstract]
Esbenshade AJ, Lu L, Friedman DL, et al.: Accumulation of Chronic Disease Among Survivors of Childhood Cancer Predicts Early Mortality. J Clin Oncol 41 (20): 3629-3641, 2023. [PUBMED Abstract]
Ehrhardt MJ, Williams AM, Liu Q, et al.: Cumulative burden of chronic health conditions among adolescent and young adult survivors of childhood cancer: Identification of vulnerable groups at key medical transitions. Pediatr Blood Cancer 68 (6): e29030, 2021. [PUBMED Abstract]
Vuotto SC, Krull KR, Li C, et al.: Impact of chronic disease on emotional distress in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 123 (3): 521-528, 2017. [PUBMED Abstract]
Hudson MM, Oeffinger KC, Jones K, et al.: Age-dependent changes in health status in the Childhood Cancer Survivor cohort. J Clin Oncol 33 (5): 479-91, 2015. [PUBMED Abstract]
Liu Q, Leisenring WM, Ness KK, et al.: Racial/Ethnic Differences in Adverse Outcomes Among Childhood Cancer Survivors: The Childhood Cancer Survivor Study. J Clin Oncol 34 (14): 1634-43, 2016. [PUBMED Abstract]
Ehrhardt MJ, Bhakta N, Liu Q, et al.: Absence of Basal Cell Carcinoma in Irradiated Childhood Cancer Survivors of Black Race: A Report from the St. Jude Lifetime Cohort Study. Cancer Epidemiol Biomarkers Prev 25 (9): 1356-60, 2016. [PUBMED Abstract]
Turcotte LM, Liu Q, Yasui Y, et al.: Temporal Trends in Treatment and Subsequent Neoplasm Risk Among 5-Year Survivors of Childhood Cancer, 1970-2015. JAMA 317 (8): 814-824, 2017. [PUBMED Abstract]
Gibson TM, Mostoufi-Moab S, Stratton KL, et al.: Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970-99: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol 19 (12): 1590-1601, 2018. [PUBMED Abstract]
Ness KK, Hudson MM, Jones KE, et al.: Effect of Temporal Changes in Therapeutic Exposure on Self-reported Health Status in Childhood Cancer Survivors. Ann Intern Med 166 (2): 89-98, 2017. [PUBMED Abstract]
Armstrong GT, Chen Y, Yasui Y, et al.: Reduction in Late Mortality among 5-Year Survivors of Childhood Cancer. N Engl J Med 374 (9): 833-42, 2016. [PUBMED Abstract]
Holmqvist AS, Chen Y, Wu J, et al.: Late mortality after autologous blood or marrow transplantation in childhood: a Blood or Marrow Transplant Survivor Study-2 report. Blood 131 (24): 2720-2729, 2018. [PUBMED Abstract]
Bagnasco F, Caruso S, Andreano A, et al.: Late mortality and causes of death among 5-year survivors of childhood cancer diagnosed in the period 1960-1999 and registered in the Italian Off-Therapy Registry. Eur J Cancer 110: 86-97, 2019. [PUBMED Abstract]
Byrne J, Schmidtmann I, Rashid H, et al.: Impact of era of diagnosis on cause-specific late mortality among 77 423 five-year European survivors of childhood and adolescent cancer: The PanCareSurFup consortium. Int J Cancer 150 (3): 406-419, 2022. [PUBMED Abstract]
Kilsdonk E, van Dulmen-den Broeder E, van Leeuwen FE, et al.: Late Mortality in Childhood Cancer Survivors according to Pediatric Cancer Diagnosis and Treatment Era in the Dutch LATER Cohort. Cancer Invest 40 (5): 413-424, 2022. [PUBMED Abstract]
Dixon SB, Liu Q, Chow EJ, et al.: Specific causes of excess late mortality and association with modifiable risk factors among survivors of childhood cancer: a report from the Childhood Cancer Survivor Study cohort. Lancet 401 (10386): 1447-1457, 2023. [PUBMED Abstract]
Mertens AC, Yong J, Dietz AC, et al.: Conditional survival in pediatric malignancies: analysis of data from the Childhood Cancer Survivor Study and the Surveillance, Epidemiology, and End Results Program. Cancer 121 (7): 1108-17, 2015. [PUBMED Abstract]
Fidler MM, Reulen RC, Winter DL, et al.: Long term cause specific mortality among 34 489 five year survivors of childhood cancer in Great Britain: population based cohort study. BMJ 354: i4351, 2016. [PUBMED Abstract]
Holmqvist AS, Chen Y, Wu J, et al.: Assessment of Late Mortality Risk After Allogeneic Blood or Marrow Transplantation Performed in Childhood. JAMA Oncol 4 (12): e182453, 2018. [PUBMED Abstract]
Dixon SB, Chen Y, Yasui Y, et al.: Reduced Morbidity and Mortality in Survivors of Childhood Acute Lymphoblastic Leukemia: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 38 (29): 3418-3429, 2020. [PUBMED Abstract]
Turcotte LM, Whitton JA, Leisenring WM, et al.: Chronic conditions, late mortality, and health status after childhood AML: a Childhood Cancer Survivor Study report. Blood 141 (1): 90-101, 2023. [PUBMED Abstract]
Ehrhardt MJ, Liu Q, Dixon SB, et al.: Association of Modifiable Health Conditions and Social Determinants of Health With Late Mortality in Survivors of Childhood Cancer. JAMA Netw Open 6 (2): e2255395, 2023. [PUBMED Abstract]
Chen C, Qin N, Wang M, et al.: Cancer germline predisposing variants and late mortality from subsequent malignant neoplasms among long-term childhood cancer survivors: a report from the St Jude Lifetime Cohort and the Childhood Cancer Survivor Study. Lancet Oncol 24 (10): 1147-1156, 2023. [PUBMED Abstract]
Anderson C, Smitherman AB, Nichols HB: Conditional relative survival among long-term survivors of adolescent and young adult cancers. Cancer 124 (14): 3037-3043, 2018. [PUBMED Abstract]
Anderson C, Nichols HB: Trends in Late Mortality Among Adolescent and Young Adult Cancer Survivors. J Natl Cancer Inst 112 (10): 994-1002, 2020. [PUBMED Abstract]
Suh E, Stratton KL, Leisenring WM, et al.: Late mortality and chronic health conditions in long-term survivors of early-adolescent and young adult cancers: a retrospective cohort analysis from the Childhood Cancer Survivor Study. Lancet Oncol 21 (3): 421-435, 2020. [PUBMED Abstract]
Rossetti S, Juul SJ, Eriksson F, et al.: Long-term cause-specific mortality in adolescent and young adult Hodgkin lymphoma patients treated with contemporary regimens-A nationwide Danish cohort study. Br J Haematol 205 (4): 1374-1382, 2024. [PUBMED Abstract]
Armenian SH, Xu L, Cannavale KL, et al.: Cause-specific mortality in survivors of adolescent and young adult cancer. Cancer 126 (10): 2305-2316, 2020. [PUBMED Abstract]
Chao C, Bhatia S, Xu L, et al.: Chronic Comorbidities Among Survivors of Adolescent and Young Adult Cancer. J Clin Oncol 38 (27): 3161-3174, 2020. [PUBMED Abstract]
Hudson MM, Armenian SH, Armstrong GT, et al.: Optimization of Health and Extension of Lifespan Through Childhood Cancer Survivorship Research. J Clin Oncol 36 (21): 2133-2134, 2018. [PUBMED Abstract]
Kremer LC, Mulder RL, Oeffinger KC, et al.: A worldwide collaboration to harmonize guidelines for the long-term follow-up of childhood and young adult cancer survivors: a report from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Pediatr Blood Cancer 60 (4): 543-9, 2013. [PUBMED Abstract]
Dixon SB, Bjornard KL, Alberts NM, et al.: Factors influencing risk-based care of the childhood cancer survivor in the 21st century. CA Cancer J Clin 68 (2): 133-152, 2018. [PUBMED Abstract]
Kirchhoff AC, Leisenring W, Krull KR, et al.: Unemployment among adult survivors of childhood cancer: a report from the childhood cancer survivor study. Med Care 48 (11): 1015-25, 2010. [PUBMED Abstract]
Mitby PA, Robison LL, Whitton JA, et al.: Utilization of special education services and educational attainment among long-term survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. Cancer 97 (4): 1115-26, 2003. [PUBMED Abstract]
Lan T, Wang M, Ehrhardt MJ, et al.: Adherence to healthy diet and risk of cardiovascular disease in adult survivors of childhood cancer in the St. Jude Lifetime Cohort: a cross-sectional study. BMC Med 21 (1): 242, 2023. [PUBMED Abstract]
Barlow-Krelina E, Chen Y, Yasui Y, et al.: Consistent Physical Activity and Future Neurocognitive Problems in Adult Survivors of Childhood Cancers: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 38 (18): 2041-2052, 2020. [PUBMED Abstract]
Tonorezos ES, Ford JS, Wang L, et al.: Impact of exercise on psychological burden in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 125 (17): 3059-3067, 2019. [PUBMED Abstract]
Lown EA, Hijiya N, Zhang N, et al.: Patterns and predictors of clustered risky health behaviors among adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 122 (17): 2747-56, 2016. [PUBMED Abstract]
Gibson TM, Liu W, Armstrong GT, et al.: Longitudinal smoking patterns in survivors of childhood cancer: An update from the Childhood Cancer Survivor Study. Cancer 121 (22): 4035-43, 2015. [PUBMED Abstract]
Devine KA, Mertens AC, Whitton JA, et al.: Factors associated with physical activity among adolescent and young adult survivors of early childhood cancer: A report from the childhood cancer survivor study (CCSS). Psychooncology 27 (2): 613-619, 2018. [PUBMED Abstract]
Mueller EL, Park ER, Kirchhoff AC, et al.: Insurance, chronic health conditions, and utilization of primary and specialty outpatient services: a Childhood Cancer Survivor Study report. J Cancer Surviv 12 (5): 639-646, 2018. [PUBMED Abstract]
Yan AP, Chen Y, Henderson TO, et al.: Adherence to Surveillance for Second Malignant Neoplasms and Cardiac Dysfunction in Childhood Cancer Survivors: A Childhood Cancer Survivor Study. J Clin Oncol 38 (15): 1711-1722, 2020. [PUBMED Abstract]
Casillas J, Oeffinger KC, Hudson MM, et al.: Identifying Predictors of Longitudinal Decline in the Level of Medical Care Received by Adult Survivors of Childhood Cancer: A Report from the Childhood Cancer Survivor Study. Health Serv Res 50 (4): 1021-42, 2015. [PUBMED Abstract]
Ford JS, Tonorezos ES, Mertens AC, et al.: Barriers and facilitators of risk-based health care for adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 126 (3): 619-627, 2020. [PUBMED Abstract]
Casillas J, Castellino SM, Hudson MM, et al.: Impact of insurance type on survivor-focused and general preventive health care utilization in adult survivors of childhood cancer: the Childhood Cancer Survivor Study (CCSS). Cancer 117 (9): 1966-75, 2011. [PUBMED Abstract]
Keegan TH, Tao L, DeRouen MC, et al.: Medical care in adolescents and young adult cancer survivors: what are the biggest access-related barriers? J Cancer Surviv 8 (2): 282-92, 2014. [PUBMED Abstract]
Kirchhoff AC, Lyles CR, Fluchel M, et al.: Limitations in health care access and utilization among long-term survivors of adolescent and young adult cancer. Cancer 118 (23): 5964-72, 2012. [PUBMED Abstract]
Nathan PC, Huang IC, Chen Y, et al.: Financial Hardship in Adult Survivors of Childhood Cancer in the Era After Implementation of the Affordable Care Act: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (5): 1000-1010, 2023. [PUBMED Abstract]
Kirchhoff AC, Kuhlthau K, Pajolek H, et al.: Employer-sponsored health insurance coverage limitations: results from the Childhood Cancer Survivor Study. Support Care Cancer 21 (2): 377-83, 2013. [PUBMED Abstract]
Kuhlthau KA, Nipp RD, Shui A, et al.: Health insurance coverage, care accessibility and affordability for adult survivors of childhood cancer: a cross-sectional study of a nationally representative database. J Cancer Surviv 10 (6): 964-971, 2016. [PUBMED Abstract]
Park ER, Kirchhoff AC, Zallen JP, et al.: Childhood Cancer Survivor Study participants’ perceptions and knowledge of health insurance coverage: implications for the Affordable Care Act. J Cancer Surviv 6 (3): 251-9, 2012. [PUBMED Abstract]
Warner EL, Park ER, Stroup A, et al.: Childhood cancer survivors’ familiarity with and opinions of the Patient Protection and Affordable Care Act. J Oncol Pract 9 (5): 246-50, 2013. [PUBMED Abstract]
Jacobs LA, Shulman LN: Follow-up care of cancer survivors: challenges and solutions. Lancet Oncol 18 (1): e19-e29, 2017. [PUBMED Abstract]
Gibson TM, Li C, Armstrong GT, et al.: Perceptions of future health and cancer risk in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 124 (16): 3436-3444, 2018. [PUBMED Abstract]
Landier W, Bhatia S, Eshelman DA, et al.: Development of risk-based guidelines for pediatric cancer survivors: the Children’s Oncology Group Long-Term Follow-Up Guidelines from the Children’s Oncology Group Late Effects Committee and Nursing Discipline. J Clin Oncol 22 (24): 4979-90, 2004. [PUBMED Abstract]
Eshelman D, Landier W, Sweeney T, et al.: Facilitating care for childhood cancer survivors: integrating children’s oncology group long-term follow-up guidelines and health links in clinical practice. J Pediatr Oncol Nurs 21 (5): 271-80, 2004 Sep-Oct. [PUBMED Abstract]
Landier W, Armenian SH, Lee J, et al.: Yield of screening for long-term complications using the children’s oncology group long-term follow-up guidelines. J Clin Oncol 30 (35): 4401-8, 2012. [PUBMED Abstract]
Wasilewski-Masker K, Mertens AC, Patterson B, et al.: Severity of health conditions identified in a pediatric cancer survivor program. Pediatr Blood Cancer 54 (7): 976-82, 2010. [PUBMED Abstract]
Subsequent Neoplasms
Subsequent neoplasms (SNs) are defined as histologically distinct neoplasms developing at least 2 months after completion of treatment for the primary malignancy. SNs may be benign or malignant. Childhood cancer survivors have increased risks of developing SNs that are multifactorial in etiology and vary according to the following:
SNs are the leading cause of nonrelapse late mortality (standardized mortality ratio, 15.2; 95% confidence interval [CI], 13.9–16.6).[1] The Childhood Cancer Survivor Study (CCSS) reported the following 30-year cumulative incidence rates:[2]
All SNs: 20.5% (95% CI, 19.1%–21.8%).
Nonmelanoma skin cancer (NMSC): 9.1% (95% CI, 8.1%–10.1%).
SNs with malignant histologies (excluding NMSC): 7.9% (95% CI, 7.2%–8.5%).
Meningioma: 3.1% (95% CI, 2.5%–3.8%).
This represents a sixfold increased risk of SNs among cancer survivors, compared with the general population.[2]
Several studies have described the excess risk of SNs.[3,4]
Evidence (excess risk of SNs):
A population-based study that leveraged registry data evaluated early second primary tumors occurring within 5 years of a first primary cancer diagnosed before age 15 years (1971–2010).[5]
Early second primary tumors developed in 0.4% of cancer survivors, which is a sevenfold excess risk (standardized incidence ratio [SIR], 7.7; 95% CI, 6.7–8.9).
Excess risk was higher among children diagnosed between 1981 and 1990 (SIR, 9.5; 95% CI, 7.1–12.5), compared with earlier or later decades (SIR, 6.5–7.5).
Cancer predisposition syndromes were implicated in 21% of children with early second primary tumors and suspected in another 5%.
An international case-control study pooled data for an analysis of meningioma risk after treatment for childhood cancer. The study evaluated the magnitude of radiation dose–response association, potential modifiers of radiation risks, and the role of chemotherapy.[6]
Increasing radiation dose was associated with increased risk of meningioma (excess odds ratio [OR]/Gy, 1.44; 95% CI, 0.6–3.6), without evidence of departure from linearity.
Exposure to radiation doses of 24 Gy or higher was associated with more than 30-fold higher odds of developing a meningioma (OR, 33.7; 95% CI, 14.1–80.3).
Patients aged 10 years or older at treatment had a significantly lower radiation dose–response association compared with those treated before the age of 10 years (excess OR/Gy, 0.57; 95% CI, 0.18–1.91 vs. 2.20; 95% CI, 0.87–6.31).
Treatment with methotrexate was associated with an increased risk of meningioma (OR, 3.43; 95% CI, 1.56–7.57), but without evidence of a dose-response association or interaction with radiation dose.
Meningioma risk associated with radiation exposure remained significantly elevated 30 years after treatment (excess OR/Gy, 3.76; 95% CI, 0.77–29.15).
A CCSS cohort reported on any new SN (including malignant neoplasms, NMSCs, benign meningiomas, and other benign neoplasms) occurring after age 40 years.[3]
At the age of 55 years, the cumulative incidence of any new SN was 34.6%. The incidence of malignant SNs was 16.3%.
Female sex and therapeutic radiation exposure were associated with an increased risk of developing SNs in multivariate analysis.
Prolonged follow-up has established that multiple SNs are common among aging childhood cancer survivors.[7,8]
Individuals treated in more recent treatment eras experienced decreased risk of SNs (including subsequent malignancies, NMSCs, and benign meningiomas) compared with those treated earlier. This lower risk was attributed to decreased exposure to therapeutic radiation.
However, individuals treated in the 1990s remain at increased risk of SNs compared with the general population.
CCSS investigators evaluated morbidity and mortality associated with meningioma among 4,221 participants treated with cranial radiation therapy.[9]
The cumulative incidence of subsequent meningioma by age 40 years was 5.6% in this group of patients, and the incidence was without demonstrable plateau.
Risk factors for subsequent meningioma included female sex (hazard ratio [HR], 1.7; 95% CI, 1.2–2.3) and higher cranial radiation dose (HR, 2.6; 95% CI, 1.6–4.2 after 30 Gy or higher).
Among survivors reporting meningiomas, the risk of neurological sequelae occurring 5 or more years after primary cancer diagnosis was increased for seizures (HR, 10.0; 95% CI, 7.0–15.3); auditory-vestibular-visual sensory deficits (HR, 2.3; 95% CI, 1.3–4.0); focal neurological dysfunction (HR, 4.9; 95% CI, 3.2–7.5); and severe headaches (HR, 3.2; 95% CI, 1.9–5.4).
With a median follow-up of 72 months after meningioma diagnosis, 13% of the patients had died, with six deaths attributed to meningioma.
Dutch Childhood Oncology Group (DCOG)–LATER investigators evaluated the cumulative meningioma incidence and performed excess relative risk modeling of benign meningiomas in childhood cancer patients.[10]
Among 5,843 childhood cancer survivors (median follow-up, 23.3 years; range, 5.0–52.2 years), 97 patients developed a benign meningioma, including 80 after full-volume cranial radiation and 14 after partial-volume cranial radiation.
The 40-year cumulative incidence of meningioma after any cranial radiation was 12.4% (95% CI, 9.8%–15.2%).
Compared with cranial radiation doses of 1 to 19 Gy, no cranial radiation was associated with a low meningioma risk (HR, 0.04; 95% CI, 0.01–0.15), while increased risks were observed for doses of 20 to 39 Gy (HR, 1.66; 95% CI, 0.83–3.33) and 40 Gy or higher (HR, 2.81; 95% CI, 1.30–6.08).
Survivors diagnosed before the age of 5 years showed significantly increased risks (HR, 2.38; 95% CI, 1.39–4.07) compared with patients diagnosed between the ages of 10 and 17 years.
Radiation volume was not significantly associated with increased risk (HR for full vs. partial volume, 1.66; 95% CI, 0.86–3.22) and dose effects did not vary significantly according to exposure age or radiation volume.
Treatment with carboplatin was associated with meningioma risk (HR, 3.55; 95% CI, 1.62–7.78), but a dose-response was not observed, and all nine exposed cases had received high-dose cranial radiation.
In a follow-up study from the DCOG-LATER cohort, 90% of survivors with meningioma presented with symptoms rather than through late effects clinic screening, and 32% of survivors presented with synchronous meningiomas.[11]
CCSS investigators have also evaluated associations between chemotherapy and subsequent malignant neoplasms (SMNs) among nonirradiated, long-term survivors.[12]
Of 1,498 SMNs in 1,344 survivors, 229 occurred in 206 survivors who were treated with chemotherapy only.
The 30-year SMN cumulative incidence was 3.9% for the chemotherapy-only group, 9.0% for the chemotherapy-plus-radiation group, 10.8% for the radiation-only group, and 3.4% for the neither-treatment group.
SIRs for SMNs were increased for any SMN (SIR, 2.8), subsequent leukemia/lymphoma (SIR, 1.9), breast cancer (SIR, 4.6), soft tissue sarcoma (SIR, 3.4), thyroid cancer (SIR, 3.8), and melanoma (SIR, 2.3).
The SMN rate was significantly associated with exposure to platinum chemotherapy doses higher than 750 mg/m2 (relative rate, 2.7) and alkylating agents (relative rate, 1.2 per 5,000 mg/m2).
The breast cancer rate showed a linear dose response (relative rate, 1.3 per 100 mg/m2) with anthracycline exposure.
DCOG-LATER investigators evaluated the contribution of chemotherapy to solid cancer risk in a large cohort of childhood cancer survivors diagnosed between 1963 and 2001 (median follow-up, 20.7 years).[13]
The 25-year cumulative SN incidence was 3.9% and did not change across decades.
Survivors treated with doxorubicin exhibited a dose-dependent increased risk of all solid cancers and breast cancer. This relationship was stronger in survivors with Li-Fraumeni syndrome–associated childhood cancers (leukemia, central nervous system [CNS], and sarcomas other than Ewing) than in survivors of other cancers.
Among female survivors who did not receive chest radiation or total-body irradiation (TBI) and developed breast cancer (n = 31), HRs for doxorubicin dose tertiles were 1.3 (95% CI, 0.3–6.1), 5.6 (95% CI, 1.9–16.2), and 9.9 (95% CI, 4.2–23.8).
A dose-response relationship was confirmed with cyclophosphamide and subsequent sarcoma, particularly bone sarcoma. The HR for subsequent sarcoma was 3.1 (95% CI, 1.5–6.0) for survivors who received cyclophosphamide at a dose greater than 9,400 mg/m2 and 2.6 (95% CI, 1.3–5.2) for those who received ifosfamide.
St. Jude Life (SJLIFE) cohort study investigators assessed the contribution of pathogenic and likely pathogenic variants in cancer predisposition genes to SN risk in childhood cancer survivors.[14]
Of 3,006 study participants evaluated by whole-genome sequencing (30-fold), 1,120 SNs were diagnosed among 439 survivors (14.6%), and 175 pathogenic or likely pathogenic variants were identified in 5.8% of survivors. The prevalence of a pathogenic or likely pathogenic variant among nonirradiated survivors with SNs was much higher, at 18%.
Variants were associated with significantly increased rates of breast cancer (relative risk [RR], 13.9) and sarcoma (RR, 10.6) among irradiated survivors and with developing any SN (RR, 4.7), breast cancer (RR, 7.7), nonmelanoma skin cancer (RR, 11), and two or more histologically distinct SNs (RR, 18.6).
Variant carriers did not have an increased rate of meningioma or thyroid cancer.
The incidence and type of SNs depend on the following:
Primary cancer diagnosis.
Type of therapy received.
Presence of genetic conditions.
Unique associations with specific therapeutic exposures have resulted in the classification of SNs into the following two distinct groups:
Myelodysplastic syndrome postcytotoxic therapy (MDS-pCT) and acute myeloid leukemia postcytotoxic therapy (AML-pCT) (previously termed t-MDS and t-AML).
Therapy-related solid SNs.
Myelodysplastic Syndrome and Acute Myeloid Leukemia Postcytotoxic Therapy (MDS-pCT and AML-pCT)
Subsequent primary leukemias have been reported in survivors of Hodgkin lymphoma, leukemia, sarcoma, CNS tumors, non-Hodgkin lymphoma, neuroblastoma, and Wilms tumor. In a cohort of nearly 70,000 5-year childhood cancer survivors, survivors had a fourfold increased risk (SIR, 3.7) of developing a leukemia, with an absolute excess risk of 7.5. Specifically, a sixfold relative risk of developing a myeloid leukemia (SIR, 5.8) was reported.[15]
The risk remained significantly elevated beyond 20 years from the first primary malignancy (SIR, 2.4).
Hodgkin lymphoma survivors had the greatest excess risk of developing a subsequent leukemia (SIR, 7.9) and specifically a myeloid leukemia (SIR, 12.1), with the largest excess risk seen before 20 years of follow-up.
A pooled analysis examined all published studies with detailed treatment data for children with cancer diagnosed between 1930 and 2000. Treatment data included estimated radiation doses to the active bone marrow and doses of specific chemotherapy agents. In this report, 147 cases of second primary leukemia (69% of cases were AML) were matched to 522 controls.[16]
After controlling for other therapies, topoisomerase II inhibitor therapy was associated with an increased risk of second AML (no radiation and >2,500 mg/m2 vs. none: OR, 14.3; 95% CI, 2.7–75.1).
Radiation dose to the active bone marrow was also associated with an increased risk of second AML among patients who did not receive chemotherapy (>0–12 Gy: OR, 4.9; 95% CI, 0.95–25.6) but not among those who received chemotherapy.
Second primary leukemias were most likely to occur in the first decade after cancer treatment.[17]
Characteristics of MDS-pCT and AML-pCT include the following:[18,19]
A short latency (<10 years from primary cancer diagnosis). The risk of MDS-pCT or AML-pCT plateaus after 10 to 15 years. Although the risk of subsequent leukemia remains significantly elevated beyond 15 years from primary diagnosis (SIR, 3.5; 95% CI, 1.9–6.0), these events are relatively rare, with an absolute excess risk of 0.02 cases per 1,000 person-years.[19]
An association with alkylating agents and/or topoisomerase II inhibitors.
Based on the updated definitions from the World Health Organization, MDS-pCT and AML-pCT are clonal disorders, which arise in patients previously exposed to cytotoxic therapy, either chemotherapy or large-field radiation therapy, for an unrelated neoplasm.[20] The following two types of MDS-pCT and AML-pCT are the most frequently observed among survivors:
Alkylating agent–related type: Alkylating agents associated with MDS-pCT and AML-pCT include cyclophosphamide, ifosfamide, mechlorethamine, melphalan, busulfan, nitrosoureas, chlorambucil, and dacarbazine.[21]
The risk of alkylating agent–related MDS or AML is dose dependent, with a latency of 3 to 5 years after exposure; it is associated with abnormalities involving chromosomes 5 (-5/del(5q)) and 7 (-7/del(7q)).[21]
Topoisomerase II inhibitor–related type: Topoisomerase II inhibitor agents include etoposide, teniposide, and anthracycline-related drugs.
Most of the translocations observed in patients exposed to topoisomerase II inhibitors disrupt a breakpoint cluster region between exons 5 and 11 of the band 11q23 and fuse KMT2A with a partner gene.[21] Topoisomerase II inhibitor–related AML presents as overt leukemia after a latency of 6 months to 3 years and is associated with balanced translocations involving chromosome bands 11q23 or 21q22.[22]
Therapy-related solid SNs represent 80% of all SNs, demonstrate a strong relationship with radiation exposure, and are characterized by a latency that exceeds 10 years. The risk of solid SNs continues to increase with longer follow-up. The risk of solid SNs is highest when the following occur:[4,13]
Younger age at time of radiation exposure.
High total dose of radiation.
Longer period of follow-up after radiation exposure.
The histological subtypes of solid SNs encompass a neoplastic spectrum ranging from benign and low-grade malignant lesions (e.g., NMSC, meningiomas) to high-grade malignancies (e.g., breast cancers, glioblastomas) (see Figure 4).[4,13,23,24]
Benign and low-grade SNs, including NMSCs and meningiomas, have also been observed with increasing prevalence in survivors who were treated with radiation therapy for childhood cancer.[4,10,30]
Subsequent neoplasms after hematopoietic stem cell transplant (HSCT)
Recipients of HSCT are treated with high-dose chemotherapy and, often, TBI, which makes their risk of SNs unique from that of the general oncology population.
Among 4,318 first-time allogeneic HSCT recipients treated for AML and chronic myeloid leukemia between 1986 and 2005, and conditioned with high-dose busulfan and cyclophosphamide (Bu-Cy), 66 solid cancers were reported at a median of 6 years post-HSCT.[31]
The cumulative incidence of new solid cancers was 0.9% at 5 years and 2.4% at 10 years and appears to be similar, regardless of exposure to radiation.
Bu-Cy conditioning without TBI was associated with higher risks of solid SNs than in the general population.
Chronic graft-versus-host disease increased the risk of SNs, especially those involving the oral cavity.
A study of 4,905 1-year survivors of allogeneic HSCT who underwent transplant between 1969 and 2014 for malignant or nonmalignant diseases, and were followed for a median 12.5 years, demonstrated a strong effect of TBI dose and dose fractionation on risk of SNs.[32]
The 20-year cumulative incidence of SN after HSCT for individuals treated at younger than 20 years was 8.1%.
SN risk was highest in survivors exposed to high-dose, single-fraction TBI (6–12 Gy) or very high-dose fractionated TBI (14.4–17.5 Gy).
With low-dose TBI (2–4.5 Gy), the SN risk was comparable to the risk with chemotherapy alone, although it was still twofold higher than in the general population.
Among individuals treated at younger than 20 years, the number of SNs was 12.5-fold higher than expected in the general population, and the excess absolute risk was 10.6 per 1,000 person-years. Survivors treated with HSCT at this young age were more likely to develop SNs than were survivors who were treated after age 50 years (HR, 2.3).
Some well-established solid SNs are described in the following sections.
Breast cancer
Female survivors of childhood, adolescent, and young adult cancer treated with radiation therapy to fields including the chest are at increased risk of developing breast cancer.
The cumulative breast cancer incidence ranges from 8% to 20% by age 40 to 45 years among childhood cancer survivors and is as high as 35% by age 50 years in Hodgkin lymphoma survivors, comparable to that observed among BRCA gene variant carriers.[18,33–36]
Radiation dose and volume of breast exposed are important factors affecting risk. Specific chemotherapeutic agents, particularly alkylating agents and anthracyclines, may affect risk as well.[33,35,37]
Evidence (excess risk of breast cancer):
Breast cancer is the most common therapy-related solid SN after a previous diagnosis of Hodgkin lymphoma.[18,34] The following has been observed in female survivors of childhood Hodgkin lymphoma:
Excess risk of breast cancer has been reported in survivors treated with high-dose, extended-volume radiation at age 30 years or younger.[38]
Data indicate that females treated with low-dose, involved-field radiation also exhibit excess breast cancer risk.[39]
Patients who received limited volume supradiaphragmatic radiation therapy (excluding the axillae) had a significantly lower risk of subsequent breast cancers than patients who received full mantle-field radiation therapy.[37]
For patients treated with radiation therapy to the chest before age 16 years, the cumulative incidence of breast cancer approaches 20% by age 45 years.[18]
The latency period after chest irradiation ranges from 8 to 10 years, and the risk of subsequent breast cancer increases in a linear fashion with radiation dose (P for trend < .001).[40]
Treatment with higher cumulative doses of alkylating agents and ovarian radiation of 5 Gy or higher (exposures predisposing to premature menopause) have been correlated with reductions in breast cancer risk, underscoring the potential contribution of hormonal stimulation on breast carcinogenesis.[37,41,42]
The observed number of invasive breast cancer cases among young (aged <30 years) survivors, compared with what is expected in the general population, has decreased over recent treatment eras (1970s SIR, 55.0 vs. 1990s SIR, 14.3).[36]
The risk of breast cancer was also increased in the following studies that used lower radiation doses to treat cancer that metastasized to the chest/lung (e.g., Wilms tumor, sarcoma) and exposed the breast tissue:
In 116 children in the CCSS cohort treated with 2 Gy to 20 Gy to the lungs (median, 14 Gy), the SIR for breast cancer was 43.6 (95% CI, 27.1–70.1).[35]
A report of 2,492 female participants in the National Wilms Tumor Studies 1 through 4 (1969–1995) addressed the excess risk of breast cancer.[43]
Sixteen of 369 women who received chest irradiation for metastatic Wilms tumor developed invasive breast cancer (cumulative risk at age 40 years, 14.8% [95% CI, 8.7%–24.5%]). The SIR of 27.6 (95% CI, 16.1–44.2) was based on 5,010 person-years of follow-up.
Of the 369 patients, radiation doses to the chest were less than 12 Gy in 4%, 12 Gy in 64%, 13 Gy to 15 Gy in 19%, and more than 15 Gy in 13% of patients.
For all patients who developed breast cancer (with or without chest irradiation), the median age at first breast cancer diagnosis was 34.3 years (range, 15.5–48.4) and the median time from Wilms tumor diagnosis was 27.1 years (range, 7.9–35.7).
An international collaborative study pooled individual patient data from 17,903 childhood cancer survivors. The study evaluated the dose-dependent effects of individual anthracycline agents on the development of subsequent breast cancer and interactions with chest radiation therapy. There were 782 survivors (4.4%) who developed a subsequent breast cancer.[44]
Doxorubicin was associated with a dose-dependent increase of subsequent breast cancer risk (HR per 100 mg/m2, 1.24; 95% CI, 1.18–1.31).
There was a more-than-twofold increased risk for survivors treated with 200 mg/m2 or higher cumulative doxorubicin dose, compared with no doxorubicin (HR, 2.50 for 200–299 mg/m2; HR, 2.33 for 300–399 mg/m2; HR, 2.78 for ≥400 mg/m2).
The associations were not statistically significant for daunorubicin, whereas epirubicin was associated with increased subsequent breast cancer risk (exposure yes vs. no: HR, 3.25; 95% CI, 1.59–6.63).
The HRs per 100 mg/m2 of doxorubicin were 1.11 (95% CI, 1.02–1.21) for patients treated with chest radiation therapy and 1.26 (95% CI, 1.17–1.36) for patients who did not receive chest radiation therapy.
The risk of developing breast cancer after radiation therapy and chemotherapy with anthracyclines was evaluated in the CCSS. In a nested-case control study of 271 childhood cancer survivors (diagnosed between 1970–1986) who were subsequently diagnosed with breast cancer, the combination of anthracyclines and radiation therapy to the breast was associated with increased risks of breast cancer consistent with an additive interaction.[33]
For the study group, the median age of first cancer diagnosis was 15 years and the median age at breast cancer diagnosis was 39 years.
The OR for breast cancer increased with increasing radiation dose to the breast (OR per 10 Gy, 3.9; 95% CI, 2.5–6.5) and was similar for estrogen receptor–positive and estrogen receptor–negative cancers.
The OR per 10 Gy to the breast was higher for women who received ovarian doses less than 1 Gy (OR, 6.8; 95% CI, 3.9–12.5) than for women who received ovarian doses greater than or equal to 15 Gy (OR, 1.4; 95% CI, 1.0–6.4).
The OR for breast cancer increased with cumulative anthracycline dose (OR per 100 mg/m2, 1.23; 95% CI, 1.09–1.39; P < .01 for trend).
There was an additive interaction between radiation therapy and anthracycline treatment. The OR was 19.1 (95% CI, 7.6–48.0) for the combined association of anthracycline therapy and breast radiation dose of 10 Gy or more (compared with 0 to less than 1 Gy) versus 9.6 (95% CI, 4.4–20.7) without anthracycline therapy.
Childhood cancer survivors not exposed to chest radiation also have an increased risk of breast cancer at a young age.
The SJLIFE study assessed subsequent breast cancer risk among 1,467 female cancer survivors and evaluated risk associated with anthracycline exposure. The study also evaluated whether surveillance imaging affects breast cancer outcomes.[25]
In women who received neither chest radiation nor anthracyclines, the cumulative incidence of breast cancer was 2% at age 35 years and 15% at age 50 years. For women who were treated with 250 mg/m2 or higher of anthracyclines, the rates were 7% at age 35 years and 46% at age 50 years.
Anthracycline doses of 250 mg/m2 or higher remained significantly associated with increased risk of breast cancer in models, excluding survivors with cancer predisposition gene variants, chest radiation of 10 Gy or higher, or both.
Breast cancers detected by imaging and/or prophylactic mastectomy were more likely to be in situ carcinomas, be smaller masses, have no lymph node involvement, and be treated without chemotherapy, compared with breast cancers detected by physical findings.
Dual imaging with mammography and breast magnetic resonance imaging (MRI) in this cohort was a sensitive and specific approach to identify breast cancers that require less aggressive therapy than breast cancers detected by physical findings.
A CCSS investigation examined the breast cancer risk of 3,768 female participants who did not receive chest radiation.[45]
A fourfold excess risk (SIR, 4.0; 95% CI, 3.0–5.3) of breast cancer was observed compared with rates in the general population.
Breast cancer risk was highest among survivors of sarcoma (SIR, 5.3; 95% CI, 3.6–7.8) and leukemia (SIR, 4.1; 95% CI, 2.4–6.9), for whom the cumulative incidence of breast cancer was estimated to be 5.8% and 6.3%, respectively, by age 45 years.
Treatment with alkylating agents and anthracyclines increased the risk of breast cancer in a dose-dependent manner.
CCSS investigators also examined SN risk among 7,448 participants who were treated with chemotherapy only.[12]
Breast cancer incidence was 4.6-fold greater than what would be expected in the general population (SIR, 4.6; 95% CI, 3.5–6.0).
A linear dose response was demonstrated between anthracyclines and breast cancer rate (RR, 1.3/100 mg/m2; 95% CI, 1.2–1.6).
DCOG-LATER investigators evaluated the contribution of chemotherapy to solid cancer risk in a large cohort of childhood cancer survivors diagnosed between 1963 and 2001.[13]
Survivors treated with doxorubicin exhibited a dose-dependent increased risk of breast cancer (HR, 3.1; 95% CI, 1.4–6.5 among survivors treated with anthracycline doses of 250 mg/m2 or higher).
The doxorubicin–breast cancer dose response was stronger for survivors of Li-Fraumeni–associated cancers (leukemia, CNS, and sarcomas other than Ewing) than for survivors of other cancers.
A SEER Program–based study evaluated trends in surgical management and the 5- and 10-year cumulative incidence of contralateral breast cancer among women who were treated with radiation therapy for Hodgkin lymphoma before age 30 years and diagnosed with a subsequent breast cancer between 1990 and 2016.[46]
Overall, 263 (89.2%) women presented with unilateral breast cancer, 50 of whom (19.0%) underwent breast-conserving surgery and 213 of whom (81.0%) underwent mastectomy (40.5% bilateral).
The 5-year incidence of contralateral breast cancer in women who underwent unilateral surgery was 9.4% (95% CI, 5.6%–15.4%), increasing to 20.2% (95% CI, 13.7%–29.2%) at 10 years and 29.9% (95% CI, 20.8%–41.9%) at 15 years. This finding underscores the importance of ongoing breast cancer surveillance and consideration of prophylactic mastectomy.
Breast cancer risk varies among childhood cancer survivors who are treated with chest radiation therapy, and the risk is based on multiple clinical factors. The first personalized breast cancer risk prediction model was developed and validated using multinational cohorts of female 5-year cancer survivors who were diagnosed at younger than 21 years and treated with chest irradiation (n = 2,147). The model includes current age, chest radiation field, whether chest radiation was delivered within 1 year of menarche, anthracycline exposure, age of menopause, and history of a first-degree relative with breast cancer. The model is available as an online risk calculator.[47]
Subsequent versus de novo breast cancer
Several studies have investigated the clinical characteristics of subsequent breast cancers arising in women treated with radiation therapy for childhood cancer.[48–52]
In one population-based study, radiation-induced breast cancer was noted to have more adverse clinicopathological features, as evidenced by a twofold increased risk of estrogen receptor–negative, progesterone receptor–negative breast cancer observed among 15-year Hodgkin lymphoma survivors, compared with women who had sporadic breast cancer.[48]
Other studies have observed a higher proportion of more histologically aggressive subtypes (e.g., triple-negative breast cancer) than age-matched sporadic invasive cancers.[49,50]
These findings are in contrast to other smaller, hospital-based, case-control studies of breast cancer among Hodgkin lymphoma survivors that have not identified a significant variation in hormone receptor status when compared with primary breast cancer controls. Previous studies have also not demonstrated a significant difference in overall risk of high-grade versus low-grade tumors.[51,52]
A study using SEER Program data evaluated the clinicopathological features of 321 breast cancers diagnosed in 257 women (median age, 22 years; range, 18–26 years at Hodgkin lymphoma diagnosis) who were previously treated with radiation for Hodgkin lymphoma.[53]
Women who developed breast cancer after radiation for Hodgkin lymphoma were younger than women diagnosed with breast cancer without a prior malignancy (43 vs. 60 years) and were less likely to present with ductal carcinoma in situ (0.4% vs. 14.9%).
Radiation was not associated with poorer biological features, compared with breast cancers diagnosed in similarly aged women with no prior malignancy. However, higher proportions of upper outer quadrant and smaller tumors (≤2 cm) were found.
Most tumors in survivors of Hodgkin lymphoma were hormone sensitive (nearly two-thirds were estrogen receptor–positive/HER2-negative). This finding suggests that endocrine prevention may be an option for this high-risk population.
Mortality after subsequent breast cancer
In a study of female participants in the CCSS who were subsequently diagnosed with breast cancer (n = 274) and matched to a control group of women (n = 1,095) with de novo breast cancer, the following was observed:[54]
Survivors of childhood cancer were found to have elevated mortality rates (HR, 2.2; 95% CI, 1.7–3.0), even after adjusting for breast cancer treatment.
Survivors were five times more likely to die from other health-related causes, including other SMNs and cardiovascular or pulmonary disease (HR, 5.5; 95% CI, 3.4–9.0).
The cumulative incidence of a second asynchronous breast cancer was elevated significantly compared with controls (at 5 years, 8.0% among childhood cancer survivors vs. 2.7% among controls; P < .001).
Although current evidence does not show a survival benefit from the initiation of breast cancer surveillance in women treated with radiation therapy to the chest for childhood cancer, interventions to promote detection of small and early-stage tumors may improve prognosis. Those with more limited treatment options because of previous exposure to radiation or anthracyclines may especially benefit.
In support of surveillance, SJLIFE investigators observed that breast cancers detected by imaging and/or prophylactic mastectomy were more likely to be in situ carcinomas, be smaller masses (3.3 cm mean tumor size detected by physical examination vs. 1.1 cm detected by imaging), have no lymph node involvement, and be treated without chemotherapy, compared with breast cancers detected by physical findings.[25]
Investigators used data from the CCSS and two Cancer Intervention and Surveillance Modeling Network breast cancer simulation models to estimate the clinical benefits, harms, and cost-effectiveness of breast cancer screening among childhood cancer survivors who were previously treated with chest radiation.[55]
Screening with mammography and MRI, as recommended in COG Long-Term Follow-Up guidelines, is projected to avert half of the expected breast cancer deaths among high-risk survivors.
On an annual schedule, a survivor will have an average of 4 to 5 false-positive screening results and 1 to 2 benign biopsy results over the course of their lifetime.
Because of the large survival benefits, the harm-benefit tradeoffs for survivors were found to be appropriate, resulting in more favorable harm-benefit ratios.
Another CCSS investigation quantified the association between temporal changes in cancer treatment over three decades and subsequent breast cancer risk.[36]
The cumulative incidence of breast cancer was 8.1% (95% CI, 7.3%–9.0%) by age 45 years among 11,550 female survivors (median age, 34.2 years), representing a more than sixfold excess risk (SIR, 6.6; 95% CI, 6.1–7.2), compared with age-, sex-, and calendar year–matched controls.
Concurrent with changes in therapy by decade, including reduced rates of chest and pelvic radiation therapy and increased rates of anthracycline chemotherapy exposure, and adjusting for age and age at diagnosis, the invasive breast cancer rate decreased 18% for every 5 years of primary cancer diagnosis era (rate ratio, 0.82; 95% CI, 0.74–0.90). This finding was largely associated with the reduced rate of chest radiation therapy.
Thyroid cancer
Thyroid cancer is observed after the following:[12,24,56,57]
Neck radiation therapy for Hodgkin lymphoma, acute lymphoblastic leukemia (ALL), and brain tumors.
Iodine I 131-metaiodobenzylguanidine (131I-MIBG) treatment for neuroblastoma.
TBI for HSCT.
Chemotherapy only, without therapeutic radiation.
The 25-year cumulative incidence of thyroid cancer among survivors of childhood cancer is 0.5%.[13] The risk of thyroid cancer among childhood cancer survivors is more than tenfold higher than that of the general population (SIR, 10.5; 95% CI, 9.1–12).[4] Significant modifiers of the radiation-related risk of thyroid cancer include the following:[28,58]
Female sex.
Younger age at exposure.
Longer time since exposure.
Radiation dose. A linear dose-response relationship between radiation exposure and thyroid cancer is observed up to 10 Gy, with a leveling off between 10 Gy and 30 Gy, and a decline in the OR at higher doses, especially in children younger than 10 years at treatment, suggesting a cell killing effect of the target cells at higher doses.[24,28,59]
In a Dutch case-control study, childhood cancer survivors with subsequent thyroid cancer were more likely to present with smaller tumors and bilateral tumors than the general population. Treatment outcomes were similar between subsequent and sporadic thyroid cancers.[60]
For information about detecting thyroid nodules and thyroid cancer, see the Thyroid nodules section.
CNS tumors
Subsequent CNS tumors represent a spectrum of histological subtypes, from high-grade gliomas to benign meningiomas. A comprehensive Pediatric Normal Tissue Effects in the Clinic (PENTEC) review analyzed the risk of SNs. The study reported a 10-year median latency period for the development of a malignant CNS neoplasm and a 21-year median latency period for the development of a meningioma.[61] Accurate assessment of the prevalence of low-grade and benign lesions is challenging because of the variable opinions and practices related to neuroimaging versus symptom surveillance in long-term survivors treated with cranial irradiation. Therefore, the prevalence of these tumors is likely higher than proven.
Brain tumors develop after cranial irradiation for histologically distinct brain tumors or for management of disease among ALL or non-Hodgkin lymphoma patients.[62] SIRs reported for subsequent CNS neoplasms after treatment for childhood cancer range from 8.1 to 52.3 across studies.[24]
The risk of subsequent brain tumors demonstrates a linear relationship with radiation dose.[26,61,63]
The risk of meningioma after radiation increases with radiation dose,[9,61] and in some studies is further potentiated with increased exposure to intrathecal methotrexate.[26] However, this finding has not been consistently replicated.[10]
Cavernomas have also been reported with considerable frequency after CNS irradiation but have been speculated to result from angiogenic processes as opposed to true tumorigenesis.[64–66]
The Dutch Long-Term Effects after Childhood Cancer (LATER) investigators have described the clinical characteristics of childhood cancer survivors who developed histologically confirmed meningiomas.[11]
In 6,015 childhood cancer survivors from the LATER cohort, 1,551 of whom had prior cranial radiation therapy, 93 survivors developed meningiomas.
Of these patients, 89 (95.7%) were treated with prior cranial radiation therapy. The median age at diagnosis was 31.8 years (range, 13.2–50.5 years).
Thirty of the survivors presented with synchronous meningiomas, and 84 survivors presented with symptoms. Only 16% of the meningiomas were detected in late effects clinics.
All survivors underwent surgery, and one-third (n = 31) of them also received radiation therapy. Twelve survivors had three or more surgeries for growth of residual tumor, recurrences, and new meningiomas. Although the extent of surgical resection was not described, the indications for radiation therapy during follow-up after surgical resection included residual tumor, recurrences, location, or new meningioma. During follow-up, 38 survivors (40.9%) developed new meningiomas, 22 (23.7%) had recurrences, and at least 4 died because of the meningioma.
The European PanCare Childhood and Adolescent Cancer Survivor Care and Follow-Up Studies (PanCareSurFup) investigators described findings in childhood cancer survivors who developed meningiomas or gliomas:[67]
In 69,460 survivors, 279 developed gliomas, and 761 developed meningiomas.
Gliomas and meningiomas were most frequently observed after any CNS tumor or leukemia, accounting for more than 70% of all observed gliomas and 80% of all meningiomas.
Among patients with known cranial radiation status who developed a glioma, 61% had received prior cranial radiation therapy. Among patients who developed a meningioma, 64.7% had received cranial radiation therapy.
CNS tumor survivors treated with cranial radiation therapy were at the highest risk of developing gliomas, at 27 times the risk of the general population (95% CI, 21.5–33.2). CNS tumor survivors who did not receive cranial radiation were at 8.3 times the risk of the general population (95% CI, 4.9–14.1).
Among CNS tumor survivors, the relative risk for patients who received cranial radiation therapy was 13 times the risk for patients who did not receive cranial radiation therapy (95% CI, 6.7–25.4). Among leukemia survivors, those who received cranial radiation therapy were at a 5-fold increased risk compared with those treated without cranial radiation therapy (95% CI, 2.9–9.9).
A PENTEC analysis of CNS SNs included 32 published studies of 1,035 subsequent meningiomas after previous radiation therapy in childhood cancer survivors.[61]
A significant dose-response relationship was reported for CNS subsequent meningiomas (excess relative ratio [ERR]/Gy, 0.44).
Younger age at the time of primary diagnosis was associated with a higher risk of subsequent meningiomas.
Females had a higher risk of subsequent meningiomas than males (odds ratio, 1.46; P < .0001).
Median latency time to the development of meningiomas was 21 years.
Neurological sequelae associated with meningiomas can include seizures, auditory-vestibular-visual deficits, focal neurological dysfunction, and severe headaches.[9] Despite the well-established increased risk of subsequent CNS neoplasms among childhood cancer survivors treated with cranial irradiation and the growing recognition of associated morbidity, the current literature is insufficient to evaluate the potential harms and benefits of routine screening for these lesions.[68] The decision to initiate surveillance should be shared by the cancer survivor and health care provider after carefully considering the potential harms and benefits of surveillance for CNS neoplasms, such as meningioma.
Proton radiation therapy for pediatric medulloblastoma is associated with low rates of brain stem injury and secondary malignancies. The long-term effects were reported in 178 pediatric patients with medulloblastoma who were treated with surgery, proton radiation therapy, and chemotherapy between 2002 and 2016 (median follow-up, 9.3 years).[69]
At 10 years, eight patients (4.5%) developed a secondary tumor (benign or malignant), with median follow up of 9.1 years. Three patients (1.7%) developed a secondary malignancy.
In-field second malignancies were glioblastoma (n = 2), meningioma (n = 2), and high-grade glioma (n = 1). The other malignancies were one each of ovarian fibroma, plexiform fibromyxoma of the esophagus, and papillary thyroid carcinoma.
Four patients developed brain stem injury at a median of 4.2 years. The 5-year cumulative incidence of brain stem injury was 1.1%, and the 10-year cumulative incidence was 1.9%.
Bone and soft tissue tumors
Survivors of hereditary retinoblastoma, Ewing sarcoma, and other malignant bone tumors are at a particularly increased risk of developing subsequent bone and soft tissue tumors.[70–74]
Radiation therapy is associated with a linear dose-response relationship.[75,76]
In a PENTEC investigation on subsequent malignancies, the estimated pooled ERR/Gy for subsequent sarcomas was 0.045. There was a possible biphasic dose response, with higher rates of relapse and subsequent sarcomas above 55 Gy. After 20 Gy and anthracyclines, the absolute excess risk is predicted to be 0.24% at 50 years and 0.86% at 75 years. The median latency time to the development of sarcomas was 11 years (range, 4–23 years).[61]
After adjustment for radiation therapy, treatment with alkylating agents [13,61] and anthracyclines [77] have both been linked to sarcoma, with the risk increasing with cumulative drug exposure.[77]
Soft tissue sarcomas can be of various histological subtypes, including nonrhabdomyosarcoma soft tissue sarcomas, rhabdomyosarcoma, malignant peripheral nerve sheath tumors, Ewing/primitive neuroectodermal tumors, and other rare tumor types.
Evidence (excess risk of bone and soft tissue tumors):
A nested case-control study included 228 cases and 228 matched controls within a cohort of 69,460 5-year survivors of childhood cancer. The study investigated the risks of subsequent primary bone cancer by different levels of cumulative radiation exposure and dose-response relationships according to different specific types of chemotherapy.[78]
Compared with unexposed bone tissue, the OR associated with bone tissue exposed to 1 to 4 Gy was 4.8-fold (95% CI, 1.2–19.6). The OR associated with bone tissue exposed to 5 to 9 Gy was 9.6-fold (95% CI, 2.4–37.4). The OR increased linearly with increasing doses of radiation (Ptrend < .001), up to 78-fold (95% CI, 9.2–669.9) for doses of 40 Gy or higher.
For cumulative alkylating agent doses of 10,000 to 19,999 mg/m2 and 20,000 mg/m2 or higher, the radiation-adjusted ORs were 7.1 (95% CI, 2.2–22.8) and 8.3 (95% CI, 2.8–24.4), respectively. There were independent contributions from exposure to procarbazine, ifosfamide, or cyclophosphamide.
A population-based study of 69,460 5-year survivors of cancer diagnosed before age 20 years observed the following:[71,72]
The risk of subsequent primary bone cancer was 22-fold greater than that of the general population, with an estimated 45-year cumulative incidence of 0.6%, compared with an expected rate of 0.03% in the general population.[71]
The observed excess numbers of subsequent primary bone cancer declined with both age and years from diagnosis.[71]
The risk of subsequent soft tissue sarcoma was almost 16-fold higher than the general population, with an estimated 45-year cumulative incidence of 1.4%, compared with an expected rate of 0.1%.[72]
The median time from diagnosis to occurrence of a soft tissue sarcoma was 19 years.[72]
The most commonly observed soft tissue sarcomas were leiomyosarcoma, fibromatous neoplasms, and malignant peripheral nerve sheath tumors.[72]
The SIR for subsequent fibromatous primary sarcomas decreased with increasing years from diagnosis and attained age, whereas the SIR for leiomyosarcoma and malignant peripheral nerve sheath tumors remained consistently high across all years from diagnosis and at all attained ages.[72]
The absolute excess risks of all sarcoma subtypes were generally low, except for leiomyosarcoma that followed a retinoblastoma diagnosis (absolute excess risks, 52.7 per 10,000 person-years among survivors 45 years or more from diagnosis).[72]
The risk of developing a leiomyosarcoma was 30-fold higher among survivors of childhood cancer, compared with an excess risk of 0.7 for the general population.[72]
Retinoblastoma survivors were at the highest risk (SIR, 342.9), followed by Wilms tumor survivors (SIR, 74.2).
90% of leiomyosarcomas observed after a Wilms tumor diagnosis developed within the irradiated tissue.
In a CCSS cohort, an increased risk of subsequent bone or soft tissue sarcoma was associated with radiation therapy, a primary diagnosis of sarcoma, a history of other SNs, and treatment with higher doses of anthracyclines or alkylating agents.[27]
The 30-year cumulative incidence of subsequent sarcoma in CCSS participants was 1.08% for survivors who received radiation therapy and 0.5% for survivors who did not receive radiation therapy.
Dose-risk modeling was used to study the risk of bone sarcoma in a retrospective cohort of 4,171 survivors of a childhood solid cancer treated between 1942 and 1986 (median follow-up, 26 years).[75]
Results demonstrated that the risk of bone sarcoma increased slightly up to a cumulative organ-absorbed radiation dose of 15 Gy (HR, 8.2; 95% CI, 1.6–42.9) and then rapidly increased for higher radiation doses (HR for 30 Gy or more, 117.9; 95% CI, 36.5–380.6), compared with patients not treated with radiation therapy.
The excess RR per Gy in this model was 1.77 (95% CI, 0.62–5.94).
In survivors of bilateral retinoblastoma, the most commonly observed malignant SN is sarcoma, specifically osteosarcoma.[79–81] The contribution of chemotherapy to solid malignancy carcinogenesis was highlighted in a long-term follow-up study of 906 5-year hereditary retinoblastoma survivors who were diagnosed between 1914 and 1996 and observed through 2009.[70]
Treatment with alkylating agents significantly increased risk of subsequent bone tumors (HR, 1.60; 95% CI, 1.03–2.49) and leiomyosarcoma (HR, 2.67; 95% CI, 1.22–5.85) among members of the cohort.
Leiomyosarcoma occurrence was more common after treatment with alkylating agent chemotherapy and radiation therapy compared with radiation therapy alone (5.8% vs. 1.6% at age 40 years; P = .01).
The CCSS reported the following on 105 cases and 422 matched controls in a nested case-control study of 14,372 childhood cancer survivors:[77]
Soft tissue sarcomas occurred at a median of 11.8 years (range, 5.3–31.3 years) from original diagnoses.
Any exposure to radiation was associated with increased risk of soft tissue sarcoma (OR, 4.1; 95% CI, 1.8–9.5), which demonstrated a linear dose-response relationship.
Anthracycline exposure was associated with soft tissue sarcoma risk (OR, 3.5; 95% CI, 1.6–7.7), independent of radiation dose.
In a cohort of 952 irradiated survivors of hereditary retinoblastoma diagnosed between 1914 and 2006, CCSS investigators observed that elevated bone and soft tissue sarcoma risks differed by age, location, and sex.[81]
Head and neck bone and soft tissue sarcomas were diagnosed beginning in early childhood and continued well into adulthood (60-year cumulative incidence of 6.8% and 9.3%, respectively).
Body and extremity bone sarcoma incidence flattened after adolescence (60-year cumulative incidence, 3.5%).
Body and extremity soft tissue sarcoma incidence was rare until age 30 years, when incidence rose steeply (60-year cumulative incidence, 6.6%) particularly for females (60-year cumulative incidence, 9.4%).
In a retrospective study of 160 patients with hereditary retinoblastoma who received radiation therapy, no correlation was identified between age (before or after 12 months) at which external-beam radiation therapy was given and development of subsequent malignancy.[73]
Patients with and without subsequent malignancies did not differ by RB1 variant type. Also, there was no association with variant type and location of SMN, or SMN type and age at diagnosis.
The study showed that patients who have a low penetrance variant and receive external-beam radiation therapy remain at risk of SMNs and should be cautiously monitored.
Skin cancer
Keratinocyte carcinomas (KCs) (i.e., basal cell carcinomas [BCCs] and squamous cell carcinomas [SCCs], also called nonmelanoma skin cancers [NMSCs]) are one of the most common SNs among childhood cancer survivors and show a strong association with radiation therapy.[4,82,83] Adherence to sun protection behaviors can reduce exposure to UV radiation that may exacerbate risk.
The CCSS performed a randomized, controlled, comparative effectiveness trial to test methods to improve early detection of skin cancer among survivors of childhood cancer at high risk after radiation therapy exposure. Participants were randomly assigned to the experimental arm, which included print materials in combination with mHealth strategies (text messages and use of the Advancing Survivors’ Knowledge website), or the control arm. Screening rates improved by 1.5-fold in the experimental arm. Rates of physician skin examination increased from baseline to 12 months, and rates of self-examination increased from baseline to 18 months in all three intervention groups. However, the increase in rates did not differ between the intervention groups.[84]
Evidence (excess risk of KCs):
An updated analysis of the CCSS confirmed an increased risk of KCs in participants with histories of radiation therapy. Compared with participants who did not receive radiation therapy, CCSS participants treated with radiation therapy had a 4.5-fold increased risk of KCs (95% CI, 3.5–5.9).[83]
The risk of KCs increased with higher radiation therapy doses. Per 10 Gy of radiation therapy at the KC site, the RR was similar for head/neck (1.0; 95% CI, 1.0–1.1) and extremity sites (1.0; 95% CI, 0.7–1.3). The RR was slightly higher for trunk sites (1.1; 95% CI, 1.0–1.3).
Allogeneic and autologous HSCT were associated with an increased risk of KCs (RR for allogeneic HSCT, 3.4; 95% CI, 2.4–4.8 and RR for autologous HSCT, 2.3; 95% CI, 1.2–4.2).
In 5,843 childhood cancer survivors in the DCOG-LATER cohort, investigators found that childhood cancer survivors had a 30-fold increased risk of developing BCCs.[30]
After a first BCC diagnosis, 46.7% of patients developed additional BCCs.
BCC risk was associated with any radiation therapy to the relevant radiation field (site of BCC) (HR, 14.32) and with estimated percentage of exposed skin surface area (26%–75%: HR, 1.99; 76%–100%: HR, 2.16 vs. 1%–25% exposed; Ptrend among exposed = .002).
BCC risk was not associated with prescribed radiation dose and likelihood of sun-exposed skin area.
Of all chemotherapy groups examined, only vinca alkaloids increased the BCC risk (HR, 1.54).
The occurrence of a KC as the first SN has been reported to identify a population at high risk of a future invasive malignant SN.[7]
CCSS investigators observed a cumulative incidence of a malignant neoplasm of 20.3% (95% CI, 13.0%–27.6%) at 15 years among radiation-exposed survivors who developed a KC as a first SN, compared with 10.7% (95% CI, 7.2%–14.2%) among survivors whose first SN was an invasive malignancy.
Malignant melanoma has also been reported as an SN in childhood cancer survivor cohorts, although at a much lower incidence than NMSCs.
Risk factors for malignant melanoma identified among these studies include the following:[85]
Radiation therapy (>40 Gy).
Alkylating agents.
Bleomycin.
Evidence (excess risk of melanoma):
Investigators assessed the incidence, risk factors, and outcomes for patients with melanoma in the CCSS cohort (n = 25,716).[85]
There were 160 survivors (median attained age, 33 years) who developed 177 melanomas (110 invasive, 62 in situ cutaneous, 5 ocular).
The 40-year cumulative incidence was 1.1% for all participants and 1.5% for those who received cumulative radiation doses of 40 Gy or higher. The standard incidence ratios were 2.0 when compared with the general population.
An increase in cutaneous melanomas was associated with a cumulative radiation dose of 40 Gy or higher to the site of the melanoma (HR, 2.0), cumulative cyclophosphamide equivalent dose of 20,000 mg/m2 or higher (HR, 1.9), and bleomycin exposure (HR, 2.2).
The presence of an invasive melanoma was associated with a twofold risk of death.
A systematic review that included data from 19 original studies (total N = 151,575 survivors; median follow-up of 13 years) observed an incidence of 10.8 cases of malignant melanoma per 100,000 childhood cancer survivors per year.[86]
Melanomas most frequently developed in survivors of Hodgkin lymphoma, hereditary retinoblastoma, soft tissue sarcoma, and gonadal tumors. However, the relatively small number of survivors represented in the relevant studies preclude assessment of melanoma risk among other types of childhood cancer.
Skin cancer risk after retinoblastoma
The incidence of melanoma and NMSC was evaluated in a cohort of 1,851 long-term, White survivors of retinoblastoma (1,020 hereditary and 831 nonhereditary) who were diagnosed from 1914 to 2006 and monitored through 2016.[87]
Of all patients, 33 hereditary and 7 nonhereditary survivors developed melanoma, and 26 hereditary and 9 nonhereditary survivors developed NMSC. The median age of skin cancer development was about 20 years younger for hereditary survivors than nonhereditary survivors.
Most NMSCs were on the head/neck, whereas melanomas were more broadly distributed, with patterns similar to melanoma-prone families.
At 50 years after retinoblastoma diagnosis, in hereditary survivors, the cumulative incidence of melanoma was 4.5% and the cumulative incidence of NMSC was 3.7%. In nonhereditary survivors, the cumulative incidence of melanoma was 0.7% and the cumulative incidence of NMSC was 1.5%.
Lung cancer
Among childhood cancer survivor cohorts, lung cancer represents a relatively uncommon SN. A PENTEC systematic review reported a median latency period of 25 years (range, 19–29 years) between the childhood cancer diagnosis and the development of a lung SN.[61] The ERR/Gy was 0.068, compared with background cumulative risks. After 20 Gy, the excess absolute risk was predicted to be 0.27% at 50 years. After 50 Gy, the excess absolute risk was predicted to be 0.7% at 50 years.[61]
Evidence (excess risk of lung cancer):
The 30-year cumulative incidence of lung cancer among CCSS participants was 0.16% (95% CI, 0.09%–0.23%), representing a SIR of 4.0 (95% CI, 2.9–5.4).[88]
Lung cancer risk demonstrated a dose-related association with chest radiation dose: 10 to 30 Gy (HR, 3.4; 95% CI, 1.05–11.0), more than 30 to 40 Gy (HR, 4.6; 95% CI, 1.5–14.3), and more than 40 Gy (HR, 9.1; 95% CI, 3.1–27.0).
Cumulative incidence of lung SMNs was higher among current or past smokers, compared with those who had never smoked.
The SIR for lung cancer was highest among survivors of Hodgkin lymphoma (SIR, 9.3; 95% CI, 6.2–13.4) and bone cancer (SIR, 4.4; 95% CI, 1.8–9.1).
The 25-year cumulative incidence of lung cancer among the DCOG-LATER cohort was 0.1% (95% CI, 0.0%–0.3%).[13]
Incidence was approximately fourfold higher than what would be expected in the general population (SIR, 4.3; 95% CI, 1.9–8.5).
Lung cancer has been reported after chest irradiation for Hodgkin lymphoma.[89]
The risk increases in association with longer elapsed time from diagnosis.
Smoking has been linked with the occurrence of lung cancer that develops after radiation therapy for Hodgkin lymphoma.[89]
The increase in risk of lung cancer with increasing radiation dose is greater among patients who smoke after exposure to radiation than among those who refrain from smoking (P = .04).
Gastrointestinal (GI) cancer
There is substantial evidence that childhood cancer survivors develop GI malignancies more frequently and at a younger age than the general population. This evidence supports the need for early initiation of colorectal carcinoma surveillance.[90–92]
Evidence (excess risk of GI cancer):
In a French and British cohort-nested, case-control study of childhood solid tumor survivors diagnosed before age 17 years, the risk of developing a digestive organ SN varied with therapy.[93]
The risk of GI cancer was 9.7-fold higher than in population controls.
The SNs most often involved the colon/rectum (42%), liver (24%), and stomach (19%).
A strong radiation dose-response relationship was observed, with an OR of 5.2 (95% CI, 1.7–16.0) for local radiation doses between 10 Gy to 29 Gy and 9.6 (95% CI, 2.6–35.2) for doses of 30 Gy and above, compared with survivors who had not received radiation therapy.
Chemotherapy alone and combined-modality therapy were associated with a significantly increased risk of developing a GI SN (SIR, 9.1; 95% CI, 2.3–23.6; SIR 29.0; 95% CI, 20.5–39.8, respectively).
The PanCare Childhood and Adolescent Cancer Survivor Care and Follow-Up Studies (PanCareSurFup) consortium quantified the absolute risks by radiation therapy treatment characteristics in a cohort of 69,450 5-year childhood cancer survivors in Europe. ORs were calculated from a case-control study comprising 143 subsequent colorectal cancers within the cohort of childhood cancer survivors.[92]
Survivors treated with any abdominopelvic radiation therapy were three times more likely to develop a subsequent colorectal cancer than those who did not receive radiation therapy (OR, 3.1; 95% CI, 1.4–6.6).
By age 40 years, survivors who were treated with abdominopelvic radiation therapy already have a similar risk of colorectal cancer as individuals aged 50 years in the general population when population-based colorectal cancer screening begins.
The risk of colorectal cancer increases with abdominopelvic radiation therapy dose. Survivors treated with spinal or whole-abdomen radiation therapy are at higher risk than those treated with radiation to other abdominopelvic fields.
CCSS investigators reported a 4.6-fold higher risk of GI SNs among their study participants than in the general population (95% CI, 3.4–6.1).[90]
The SNs most often involved the colon (39%), rectum/anus (16%), liver (18%), and stomach (13%).
The SIR for colorectal cancer was 4.2 (CI, 2.8–6.3).
The most prevalent GI SN histology was adenocarcinoma (56%).
The highest risk of GI SNs was associated with abdominal irradiation (SIR, 11.2; CI, 7.6–16.4), but survivors not exposed to radiation also had a significantly increased risk (SIR, 2.4; CI, 1.4–3.9).
High-dose procarbazine (RR, 3.2; CI, 1.1–9.4) and platinum drugs (RR, 7.6; CI, 2.3–25.5) independently increased the risk of GI SNs.
St. Jude Children’s Research Hospital investigators observed that the SIR for subsequent colorectal carcinoma was 10.9 (95% CI, 6.6–17.0) compared with U.S. population controls. Investigators also observed the following:[91]
Incidence of a subsequent colorectal carcinoma increased steeply with advancing attained age, with a 40-year cumulative incidence of 1.4% ± 0.53% among the entire cohort (N = 13,048) and 2.3% ± 0.83% for 5-year survivors.
Colorectal carcinoma risk increased by 70% with each 10 Gy increase in radiation dose. Increasing radiation volume also increased the risk.
Treatment with alkylating agent chemotherapy was also associated with an 8.8-fold excess risk of subsequent colorectal carcinoma.
A multi-institutional prospective study observed that potentially precancerous neoplastic polyps were found in 27.8% of childhood cancer survivors who received radiation to the abdomen/pelvis at least 10 years earlier and who had colonoscopic screening between age 35 and 49 years.[94]
This polyp prevalence is at least as high as that previously reported for the average-risk population older than 50 years and is similar to the 24% incidence rate for patients with hereditary nonpolyposis colon cancer. Polyp prevalence rates in the general population for people aged 35 to 49 years are unclear.
A DCOG-LATER record linkage study evaluated the risk of histologically confirmed colorectal adenomas among 5,843 5-year childhood cancer survivors followed for a median of 24.9 years.[95]
The cumulative incidence of colorectal adenoma by age 45 years was 3.6% among survivors who received abdominal pelvic radiation versus 2.0% for survivors who did not receive abdominal pelvic radiation, versus 1.0% among siblings.
Factors associated with adenoma risk were abdominal pelvic radiation (HR, 2.1), TBI (HR, 10.6), cisplatin (HR, 2.1 for <480 mg/m2; HR, 3.8 for ≥480 mg/m2), diagnosis of hepatoblastoma (HR, 27.1), and family history of early-onset colorectal cancer (HR, 20.5).
Procarbazine exposure was also associated with an increased risk among survivors not exposed to abdominal pelvic radiation or TBI (HR, 2.7).
The PanCareSurFup consortium reported on digestive cancers in a cohort of 69,460 5-year childhood cancer survivors in Europe.[96]
Survivors of Wilms tumor (SIR, 12.1; 95% CI, 9.6–15.1) and Hodgkin lymphoma (SIR, 7.3; 95% CI, 5.9–9.0) were at the highest risk for GI SMNs.
By age 55 years, 2.3% of survivors of Wilms tumor and Hodgkin lymphoma developed a secondary colorectal cancer, a rate that is comparable to that of the general population with two or more first-degree relatives affected by GI cancer.
A nested case-control study examined the rate of colorectal cancer according to large bowel radiation therapy dose and procarbazine dose among 5-year Hodgkin lymphoma survivors who were diagnosed between age 15 and 50 years (only 27% of cases were 15 to 24 years old) at five hospital centers in the Netherlands (diagnosed between 1964 to 2000; median follow-up, 26 years).[97]
The median interval between Hodgkin lymphoma diagnosis and subsequent colorectal cancer was 25.7 years (range, 18.2–31.6 years).
Treatment with subdiaphragmatic radiation therapy (RR, 2.4; 95% CI, 1.4–4.1) and more than 8.4 g/m2 of procarbazine (RR, 2.5; 95% CI, 1.3–5.0) were associated with increased rates of colorectal cancer on univariable analysis.
Colorectal cancer rate increased linearly with mean radiation therapy dose to the whole large bowel and dose to the affected bowel segment. The dose response became steeper with higher doses of procarbazine and increased 1.2-fold (95% CI, 1.1–1.3) for each 1 g/m2 increase in procarbazine.
Oral cancers
The PanCareSurFup consortium reported on risks of oral second primary neoplasms (validated through pathology reports) in a cohort of 69,460 5-year childhood cancer survivors in Europe.[98]
Oral second primary neoplasms (n = 145; 64 salivary gland, 38 tongue, 20 pharynx, 2 lip, and 21 other) developed in 143 survivors at a median age of 32 years. This represents a fivefold excess risk compared with that expected in the general population.
Childhood cancer diagnostic groups at greatest risk for an oral second primary neoplasm included leukemia (SIR, 19.2; 95% CI, 14.6–25.2), bone sarcoma (SIR, 6.4; 95% CI, 3.7–11.0), Hodgkin lymphoma (SIR, 6.2; 95% CI, 3.9–9.9), and soft tissue sarcoma (SIR, 5.0; 95% CI, 3.0–8.5).
Observed specific treatment associations with oral second primary neoplasms included radiation therapy and salivary gland neoplasms (SIR, 33; 95% CI, 25.3–44.5) and chemotherapy (any exposure) and tongue neoplasms (SIR, 15.9; 95% CI, 10.6–23.7).
Data were not available to assess the contributions of health behaviors (smoking, alcohol intake) and human papillomavirus (HPV) infection status to the development of oral second primary neoplasms.
Urogenital cancers
Development of subsequent primary urogenital cancers in childhood and adolescent cancer survivors is rare.
Using SEER data of 43,991 patients (aged <20 years) diagnosed with a first primary cancer from 1975 to 2016, the risk of urinary system cancer was higher for both females (SIR, 5.18; 95% CI, 3.65–7.14) and males (SIR, 2.80; 95% CI, 1.94–3.92), compared with the general population.[99]
Females were more likely than males to develop a subsequent urinary system cancer (SIR, 1.86; 95% CI, 1.13–3.03) and kidney cancer (SIR, 1.97; 95% CI, 1.11–3.53).
Females with any first cancer had higher risks than the general population for developing cancers of the corpus uteri (SIR, 2.32; 95% CI, 1.49–3.45) and vulva (SIR, 4.27; 95% CI, 1.38–9.95).
Renal carcinoma
Consistent with reports among survivors of adult-onset cancer, an increased risk of renal carcinoma has been observed in survivors of childhood cancer.[29,100,101] Underlying genetic predisposition may also play a role in the risk of developing renal carcinomas because rare cases of renal carcinoma have been observed in children with tuberous sclerosis.[100] Cases of secondary renal carcinoma associated with Xp11.2 translocations and TFE3 gene fusions have also been reported and suggest that cytotoxic chemotherapy may contribute to renal carcinogenesis.[102–104]
Evidence (excess risk of renal carcinoma):
CCSS investigators reported a significant excess of subsequent renal carcinoma among 14,358 5-year survivors in the cohort (SIR, 8.0; 95% CI, 5.2–11.7) compared with the general population.[100]
The reported overall absolute excess risk of 8.4 per 105 person-years indicates that these cases are relatively rare. Highest risk was observed among the following:
Neuroblastoma survivors (SIR, 85.8; 95% CI, 38.4–175.2).[100] Radiation has been hypothesized to predispose children with high-risk neuroblastoma to renal carcinoma.[105]
Those treated with renal-directed radiation therapy of 5 Gy or higher (RR, 3.8; 95% CI, 1.6–9.3).[100]
Those treated with platinum-based chemotherapy (RR, 3.5; 95% CI, 1.0–11.2).[100]
Human Papillomavirus (HPV)–Associated Malignancies
Evidence (HPV-associated SMNs):
A CCSS study evaluated the occurrence of cancer types in which HPV is an established etiologic risk factor. The study included 24,363 childhood cancer survivors who were a median of 21 years from diagnosis.[106]
The 30-year cumulative incidence of an HPV-associated cancer was 0.3% (95% CI, 0.2%–0.4%), reflecting an almost threefold excess risk (SIR, 2.86; 95% CI, 2.05–4.00) to that of the general population.
Male and female cancer survivors had an elevated risk of HPV-associated oropharyngeal (SIR in males, 4.06; SIR in females, 8.44) and anorectal (SIR in males, 13.56; SIR in females, 9.16) SMNs. However, females were not at increased risk of cervical or vulvar cancers, compared with the general population.
Independent risk factors for HPV-associated cancers identified by multivariable modeling included male sex (vs. females: relative SIR, 1.99; 95% CI, 1.00–3.94); head, neck, and pelvic radiation therapy doses higher than 30 Gy (vs. none: relative SIR, 2.35; 95% CI, 1.11–4.97); and cisplatin-equivalent doses higher than 400 mg/m2 (vs. none: relative SIR, 4.51; 95% CI, 1.78–11.43).
A population-based study used Surveillance, Epidemiology, and End Results (SEER) registry data to evaluate risk factors and trends for HPV-associated SMNs among 374,408 adolescent and young adult (AYA) patients (11% were aged 15–24 years, 28% were aged 25–34 years, and 61% were aged 35–44 years at diagnosis). The patients were diagnosed between 1976 and 2015.[107]
The incidence of HPV-associated SMNs declined over the study period, and the overall incidence of HPV-associated SMNs among AYA cancer survivors was low, affecting only 0.4% of survivors.
Risk of any HPV-associated SMN was increased by 70% for AYA survivors (SIR, 1.70; 95% CI, 1.61–1.79) and 117% for oropharyngeal cancer (SIR, 2.17; 95% CI, 2.00–2.35), compared with the general population.
Cervical cancer risk was overall lower in survivors (SIR, 0.85; 95% CI, 0.76–0.95), but Hispanic AYA survivors had a significant increase in cervical cancer (SIR, 1.46; 95% CI, 1.01–2.06).
Among survivors with HPV-related first cancers, prior chemotherapy and radiation therapy were associated with any HPV-related SMN. However, those therapies were not associated with HPV-related SMNs among survivors whose first cancers were not HPV related.
Survival Outcomes After SNs
Outcome after the diagnosis of an SN is variable, as treatment for some histological subtypes may be compromised if childhood cancer therapy included cumulative doses of agents and modalities at the threshold of tissue tolerance.
Using data from the SEER Program, individuals younger than 60 years with first primary malignancies (n = 1,332,203) were compared with childhood cancer survivors (n = 1,409) who had a second primary malignancy.[108]
Survivors of childhood cancer diagnosed with a second primary malignancy experienced poorer overall survival than did their peers without a history of cancer (HR, 1.86; 95% Cl, 1.72–2.02) after the study had accounted for cancer type, age, sex, race, and decade of diagnosis.
A history of childhood cancer was consistently associated with a twofold to threefold increased risk of death for the most diagnosed second primary malignancies, including breast cancer, thyroid cancer, AML, brain cancer, melanoma, bone cancer, and soft tissue sarcoma.
In a study of female participants in the CCSS who were subsequently diagnosed with breast cancer (n = 274) and matched to a control group of women (n = 1,095) with de novo breast cancer, survivors of childhood cancer were found to have elevated mortality rates (HR, 2.2; 95% CI, 1.7–3.0) even after adjusting for breast cancer treatment.[54]
Survivors were five times more likely to die as a result of other health-related causes, including other SMNs and cardiovascular or pulmonary disease (HR, 5.5; 95% CI, 3.4–9.0).
The cumulative incidence of a second asynchronous breast cancer was elevated significantly compared with controls (at 5 years, 8.0% among childhood cancer survivors vs. 2.7% among controls; P < .001).
Subsequent Neoplasms and Genetic Susceptibility
Literature clearly supports the role of chemotherapy and radiation therapy in the development of SNs. However, interindividual variability exists, suggesting that genetic variation has a role in susceptibility to genotoxic exposures, or that genetic susceptibility syndromes confer an increased risk of cancer, such as Li-Fraumeni syndrome.[109,110] In a population-based Swiss Childhood Cancer Survivor Study, cancer predisposition syndromes were associated with a high risk of second primary neoplasms before the age of 21 years and represented the most important risk factor (HR, 7.8; 95% CI, 4.8–12.7) for developing a second primary cancer.[111]
Previous studies have demonstrated that childhood cancer survivors with a family history of Li-Fraumeni syndrome in particular, or a family history of cancer, carry an increased risk of developing an SN.[112,113] A prospective registry followed 480 individuals with pathogenic or likely pathogenic germline TP53 variants.[114] Individuals who developed a first cancer were monitored for the development of a second malignant neoplasm. Among individuals who were younger than 17 years at the time of diagnosis of their first cancer, 50% developed a second cancer within 20 years.
The risk of SNs could potentially be modified by variants in high-penetrance genes that lead to these serious genetic diseases (e.g., Li-Fraumeni syndrome).[113] However, the attributable risk is expected to be very small because of the extremely low prevalence of variants in high-penetrance genes.
Likewise, children with neurofibromatosis type 1 (NF1) who develop a primary tumor are at an increased risk of SNs compared with childhood cancer survivors without NF1. Treatment with radiation, but not alkylating agents, increases the risk of SNs in survivors with NF1.[115] SNs represent a major contributor to excess mortality in adult survivors of childhood glioma with NF1.[116] These survivors developed late-onset (>5 years from diagnosis) SMNs at four times the rate of glioma survivors without NF1 (4.02; range, 2.12–7.62). The 30-year, all-cause late mortality rate was 46.3% (95% CI, 23.9%–62.2%) in glioma survivors with NF1, compared with 18% (95% CI, 16.1%–20.0%) in glioma survivors without NF1. The most common causes of death among survivors with NF1 and glioma were SNs.
Table 1 summarizes the spectrum of neoplasms, affected genes, and Mendelian mode of inheritance of selected syndromes of inherited cancer predisposition.
Table 1. Selected Syndromes of Inherited Cancer Predispositiona
Syndrome
Major Tumor Types
Affected Gene
Mode of Inheritance
AML = acute myeloid leukemia; MDS = myelodysplastic syndromes; WAGR = Wilms tumor, aniridia, genitourinary abnormalities, and range of developmental delays.
Hemangioblastoma, pheochromocytoma, renal cell carcinoma, retinal and central nervous system tumors
VHL
Dominant
WAGR syndrome
Gonadoblastoma, Wilms tumor
WT1
Dominant
Wilms tumor syndrome
Wilms tumor
WT1
Dominant
Xeroderma pigmentosum
Leukemia, melanoma
XPA, XPB, XPC, XPD, XPE, XPF, XPG, POLH
Recessive
The McGill Interactive Pediatric OncoGenetic Guidelines (MIPOGG) tool identifies children with cancer who have an increased likelihood of having a cancer predisposition syndrome. This tool guides clinicians through a series of yes or no questions, and it generates a recommendation for or against genetic evaluation.[118,119]
In a population-based, nested, case-control study, the MIPOGG tool identified survivors who were at increased risk of developing SMNs, after controlling for radiation and chemotherapy exposures (HR, 1.53; 95% CI, 1.06–2.19).
SMN prediction was superior in survivors of CNS and solid tumor neoplasms and in survivors who were not irradiated during treatment of their primary malignancy.
Use of the MIPOGG tool in pediatric oncology patients, at diagnosis or in survivorship follow-up, may help prioritize those who should undergo genetic evaluation.
Drug-metabolizing enzymes and DNA repair polymorphisms
The interindividual variability in risk of SNs is more likely related to common polymorphisms in low-penetrance genes that regulate the availability of active drug metabolites or are responsible for DNA repair. Gene-environment interactions may magnify subtle functional differences resulting from genetic variations.
In related research, SJLIFE investigators evaluated cancer treatments and pathogenic germline variants in 127 genes from six major DNA repair pathways to identify childhood cancer survivors at an increased risk of SNs.[120]
Among 4,402 survivors who underwent whole-genome sequencing, 495 (11.2%) developed 1,269 SNs.
Among 508 survivors (11.5%), 538 pathogenic germline variants were identified in 98 DNA repair pathways (e.g., POLG, MUTYH, ERCC2, and BRCA2).
The following three groups were identified to have an elevated risk of SNs:
Female survivors with variants in homologous recombination genes who were treated with high doses (≥20 Gy) of chest radiation (RR, 4.4; 95% CI, 1.6–12.4) or a cumulative dose of anthracyclines in the second or third tertile (RR, 4.4; 95% CI, 1.7–11.4) had a significantly increased risk of breast cancer.
Survivors with variants in homologous recombination genes who received alkylating agent doses in the third tertile had an increased rate of subsequent sarcomas (RR, 14.9; 95% CI, 4.0–38.0).
Survivors with variants in nucleotide excision repair genes who were treated with neck radiation (≥30 Gy) had an increased risk of subsequent thyroid cancer (RR, 12.9; 95% CI, 1.6–46.6).
Drug-metabolizing enzymes
Metabolism of genotoxic agents occurs in two phases.
Phase I involves activation of substrates into highly reactive electrophilic intermediates that can damage DNA, a reaction principally performed by the cytochrome p450 (CYP) family of enzymes.
Phase II enzymes (conjugation) function to inactivate genotoxic substrates. The phase II proteins comprise the glutathione S-transferase (GST) enzymes, NAD(P)H:quinone oxidoreductase-1 (NQO1) enzyme, and others.
The balance between the two sets of enzymes is critical to the cellular response to xenobiotics; for example, high activity of a phase I enzyme and low activity of a phase II enzyme can result in DNA damage.
DNA repair polymorphisms
DNA repair mechanisms protect somatic cells from variants in tumor suppressor genes and oncogenes that can lead to cancer initiation and progression. An individual’s DNA repair capacity appears to be genetically determined.[121] A number of DNA repair genes contain polymorphic variants, resulting in large interindividual variations in DNA repair capacity.[121] Evaluation of the contribution of polymorphisms influencing DNA repair to the risk of SN represents an active area of research.
Polygenic risk
Survivors of childhood cancer are at an increased risk of subsequent cancers attributable to the late effects of radiation therapy and other treatment exposures. To better understand the impact of genetic predisposition on this risk, several studies have examined radiation-related cancers and chemotherapy only–related cancers.
Investigators combined genotype data for 11,220 5-year survivors from the CCSS and SJLIFE cohort study. They conducted a comprehensive investigation of general population, cancer-specific polygenic risk scores (PRS) derived from genome-wide association study findings. The study aimed to identify whether the PRS were associated with subsequent cancer risk among survivors of childhood cancer, after controlling for treatment exposure and nongenetic risk factors. They also quantified joint associations between radiation therapy and PRS to understand the potential interplay between genetic and treatment risk factors.[122]
In survivors of predominantly European ancestry, standardized PRS were significantly associated with the risk of subsequent BCCs (OR, 1.37; n = 958), female breast cancers (OR, 1.42; n = 360), thyroid cancers (OR, 1.48; n = 259), squamous cell carcinomas (OR, 1.20; n = 127), and melanomas (OR, 1.60; n = 103). The association between colorectal cancer PRS and subsequent colorectal cancer risk did not reach the threshold for statistical significance (OR, 1.19; n = 67).
Childhood cancer survivors with a higher PRS and who received radiation therapy had more-than-additive increased risks of developing BCCs, breast cancers, and thyroid cancers.
For female survivors who were exposed to chest radiation therapy (>10 Gy), those with high PRS had an increased cumulative incidence of subsequent breast cancer by age 50 years, compared with those with low PRS.
With the decreased use of radiation therapy, it has become important to define the role of genetic susceptibility in chemotherapy-related SMNs. SJLIFE cohort study investigators evaluated treatment-related SMNs among long-term survivors of childhood cancer. An externally validated 179-variant PRS associated with risks of common adult-onset cancers in the general population was calculated for each survivor.[123]
The most frequent SMNs developing in 1,594 survivors included BCC (n = 822), breast cancer (n = 235), and thyroid cancer (n = 221).
SMN risk associations with the PRS were extremely modest in survivors with European ancestry who were exposed to radiation therapy (HR, 1.22; n = 4,630).
Among survivors with European ancestry who did not receive radiation therapy (n = 4,322), the increase in 30-year SMN cumulative incidence and HRs comparing top and bottom PRS quintiles was statistically significant for those treated with alkylating agents (17% vs. 6%; HR, 2.46; P < .01), anthracyclines (20% vs. 8%; HR, 2.86; P < .001), epipodophyllotoxins (23% vs. 1%; HR, 12.20; P < .001), or platinums (46% vs. 7%; HR, 8.58; P < .01).
Among survivors with African ancestry who did not receive radiation therapy (n = 414), the PRS also significantly predicted epipodophyllotoxin-related SMN risk. The greatest improvements in risk prediction were for survivors who were exposed to epipodophyllotoxins (HR, 2.68; P < .01).
Screening and Follow-Up for Subsequent Neoplasms
Vigilant screening is important for childhood cancer survivors at risk.[124] Because of the relatively small size of the pediatric cancer survivor population and the prevalence and time to onset of therapy-related complications, undertaking clinical studies to assess the impact of screening recommendations on the morbidity and mortality associated with the late effect is not feasible.
Well-conducted studies of large populations of childhood cancer survivors have provided compelling evidence linking specific therapeutic exposures and late effects. This evidence has been used by several national and international cooperative groups (Scottish Collegiate Guidelines Network, Children’s Cancer and Leukaemia Group, Children’s Oncology Group [COG], DCOG) to develop consensus-based clinical practice guidelines to increase awareness and standardize the immediate care needs of medically vulnerable childhood cancer survivors.[125]
All pediatric cancer survivor health screening guidelines employ a hybrid approach that is both evidence-based (using established associations between therapeutic exposures and late effects to identify high-risk categories) and grounded in the collective clinical experience of experts (matching the magnitude of the risk with the intensity of the screening recommendations). The screening recommendations in these guidelines represent a statement of consensus from a panel of experts in the late effects of pediatric cancer treatment.[124,125]
The COG Guidelines for malignant SNs indicate that certain high-risk populations of childhood cancer survivors merit heightened surveillance because of predisposing host, behavioral, or therapeutic factors.[124]
Screening for leukemia: MDS-pCT or AML-pCT usually manifests within 10 years after exposure. Recommendations include monitoring with history and physical examination for signs and symptoms of pancytopenia for 10 years after exposure to alkylating agents or topoisomerase II inhibitors.
Screening after radiation exposure: Most other SNs are associated with radiation exposure and usually manifest more than 10 years after exposure. Screening recommendations include careful annual physical examination of the skin and exposed (often underlying) tissues in the radiation field.
Specific comments about screening for more common radiation-associated SNs are as follows:
Screening for early-onset skin cancer: Annual dermatological examination focusing on skin lesions and pigmented nevi in the radiation field is recommended. Survivors are counseled about the following:
Increased risk of skin cancer.
Potential exacerbation of risk through tanning.
Benefits of adhering to behaviors to protect the skin from excessive UV radiation exposure.
Screening for early-onset breast cancer: Because outcome after breast cancer is directly linked to stage at diagnosis, close surveillance that results in early diagnosis may improve survival.[126] Several pediatric cancer groups have endorsed the recommendation for early (before population breast cancer screening) initiation of breast cancer surveillance using mammography, breast MRI, or both imaging modalities in young women who were treated with chest irradiation.[126]
Mammography, the most widely accepted screening tool for breast cancer in the general population, may not be the ideal screening tool by itself for radiation-related breast cancers occurring in relatively young women with dense breasts. On the basis of research among young women with inherited susceptibility to breast cancer, dual-imaging modalities may enhance early detection related to the higher sensitivity of MRI in detecting lesions in premenopausal dense breasts and the superiority of mammography in identifying ductal carcinoma in situ;[127–129] therefore, the American Cancer Society recommends including adjunct screening with MRI.[130] The high sensitivity and specificity in detecting early-stage lesions with dual-imaging surveillance is offset by a substantial rate of additional investigations attributable to false-positive results.[129]
Many clinicians are concerned about potential harms related to radiation exposure associated with annual mammography in these young women. In this regard, it is important to consider that the estimated mean breast dose with contemporary standard two-view screening mammograms is about 3.85 mGy to 4.5 mGy.[131–133] Thus, 15 additional surveillance mammograms from age 25 to 39 years would increase the total radiation exposure in a woman treated with 20 Gy of chest radiation to 20.05775 Gy. The benefits of detection of early breast cancer lesions in high-risk women must be balanced by the risk predisposed by a 0.3% additional radiation exposure.
To keep young women engaged in breast health surveillance, the COG Guideline recommends the following for females who received a radiation dose of 10 Gy or higher to the mantle, mediastinal, whole lung, and axillary fields:
Monthly breast self-examination beginning at puberty.
Annual clinical breast examinations beginning at puberty until age 25 years.
A clinical breast examination every 6 months, with annual mammograms and MRIs beginning 8 years after radiation therapy or at age 25 years (whichever occurs later).
The risk of breast cancer in patients who received less than 10 Gy of radiation with potential impact to the breast is of a lower magnitude compared with those who received 10 Gy or higher. Monitoring of patients treated with less than 10 Gy of radiation with potential impact to the breast is determined on an individual basis after a discussion with the provider regarding the benefits and risk/harms of screening. If a decision is made to screen, the recommendations for women exposed to 10 Gy or higher are used.
Screening for thyroid cancer: Surveillance recommendations for thyroid cancer among survivors treated with radiation therapy to the thyroid gland vary a great deal. Consensus recommendations for surveillance were developed by the International Guidelines Harmonization Group (IGHG) with the PanCareSurFup consortium. Ultimately, neither of the two surveillance strategies, thyroid ultrasonography and neck palpation, was demonstrated to be superior. This finding highlights the need for shared decision-making between the medical provider and patient.[134]
Screening for subsequent CNS neoplasms: The IGHG identified a paucity of high-quality evidence for CNS screening. The group determined there was insufficient evidence to establish whether early CNS neoplasm detection results in reduced morbidity and/or mortality. Therefore, a recommendation was not made for or against MRI surveillance. Shared decision-making between the medical provider and survivor is recommended to formulate a screening plan.[68]
Screening for early-onset colorectal cancer: Screening of those at risk of early-onset colorectal cancer (i.e., radiation doses of 20 Gy or higher to the abdomen, pelvis, or spine) includes colonoscopy every 5 years or multitarget stool DNA test every 3 years beginning at age 30 years or 5 years after radiation therapy (whichever occurs later). For more information, see the COG Long-Term Follow-Up Guidelines for Survivors of Childhood, Adolescent, and Young Adult Cancers.
References
Mertens AC, Liu Q, Neglia JP, et al.: Cause-specific late mortality among 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst 100 (19): 1368-79, 2008. [PUBMED Abstract]
Friedman DL, Whitton J, Leisenring W, et al.: Subsequent neoplasms in 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst 102 (14): 1083-95, 2010. [PUBMED Abstract]
Turcotte LM, Whitton JA, Friedman DL, et al.: Risk of Subsequent Neoplasms During the Fifth and Sixth Decades of Life in the Childhood Cancer Survivor Study Cohort. J Clin Oncol 33 (31): 3568-75, 2015. [PUBMED Abstract]
Turcotte LM, Liu Q, Yasui Y, et al.: Temporal Trends in Treatment and Subsequent Neoplasm Risk Among 5-Year Survivors of Childhood Cancer, 1970-2015. JAMA 317 (8): 814-824, 2017. [PUBMED Abstract]
Stiller CA, Bunch KJ, Bayne AM, et al.: Subsequent cancers within 5 years from initial diagnosis of childhood cancer. Patterns and risks in the population of Great Britain. Pediatr Blood Cancer 70 (5): e30258, 2023. [PUBMED Abstract]
Withrow DR, Anderson H, Armstrong GT, et al.: Pooled Analysis of Meningioma Risk Following Treatment for Childhood Cancer. JAMA Oncol 8 (12): 1756-1764, 2022. [PUBMED Abstract]
Armstrong GT, Liu W, Leisenring W, et al.: Occurrence of multiple subsequent neoplasms in long-term survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol 29 (22): 3056-64, 2011. [PUBMED Abstract]
van Eggermond AM, Schaapveld M, Lugtenburg PJ, et al.: Risk of multiple primary malignancies following treatment of Hodgkin lymphoma. Blood 124 (3): 319-27; quiz 466, 2014. [PUBMED Abstract]
Bowers DC, Moskowitz CS, Chou JF, et al.: Morbidity and Mortality Associated With Meningioma After Cranial Radiotherapy: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 35 (14): 1570-1576, 2017. [PUBMED Abstract]
Kok JL, Teepen JC, van Leeuwen FE, et al.: Risk of benign meningioma after childhood cancer in the DCOG-LATER cohort: contributions of radiation dose, exposed cranial volume, and age. Neuro Oncol 21 (3): 392-403, 2019. [PUBMED Abstract]
Verbruggen LC, Kok JL, Teepen JC, et al.: Clinical characteristics of subsequent histologically confirmed meningiomas in long-term childhood cancer survivors: A Dutch LATER study. Eur J Cancer 150: 240-249, 2021. [PUBMED Abstract]
Turcotte LM, Liu Q, Yasui Y, et al.: Chemotherapy and Risk of Subsequent Malignant Neoplasms in the Childhood Cancer Survivor Study Cohort. J Clin Oncol 37 (34): 3310-3319, 2019. [PUBMED Abstract]
Teepen JC, van Leeuwen FE, Tissing WJ, et al.: Long-Term Risk of Subsequent Malignant Neoplasms After Treatment of Childhood Cancer in the DCOG LATER Study Cohort: Role of Chemotherapy. J Clin Oncol 35 (20): 2288-2298, 2017. [PUBMED Abstract]
Wang Z, Wilson CL, Easton J, et al.: Genetic Risk for Subsequent Neoplasms Among Long-Term Survivors of Childhood Cancer. J Clin Oncol 36 (20): 2078-2087, 2018. [PUBMED Abstract]
Allodji RS, Hawkins MM, Bright CJ, et al.: Risk of subsequent primary leukaemias among 69,460 five-year survivors of childhood cancer diagnosed from 1940 to 2008 in Europe: A cohort study within PanCareSurFup. Eur J Cancer 117: 71-83, 2019. [PUBMED Abstract]
Allodji RS, Tucker MA, Hawkins MM, et al.: Role of radiotherapy and chemotherapy in the risk of leukemia after childhood cancer: An international pooled analysis. Int J Cancer 148 (9): 2079-2089, 2021. [PUBMED Abstract]
Ghosh T, Hyun G, Dhaduk R, et al.: Late subsequent leukemia after childhood cancer: A report from the Childhood Cancer Survivor Study (CCSS). Cancer Med 13 (20): e70086, 2024. [PUBMED Abstract]
Bhatia S, Yasui Y, Robison LL, et al.: High risk of subsequent neoplasms continues with extended follow-up of childhood Hodgkin’s disease: report from the Late Effects Study Group. J Clin Oncol 21 (23): 4386-94, 2003. [PUBMED Abstract]
Nottage K, Lanctot J, Li Z, et al.: Long-term risk for subsequent leukemia after treatment for childhood cancer: a report from the Childhood Cancer Survivor Study. Blood 117 (23): 6315-8, 2011. [PUBMED Abstract]
Khoury JD, Solary E, Abla O, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36 (7): 1703-1719, 2022. [PUBMED Abstract]
Thirman MJ, Larson RA: Therapy-related myeloid leukemia. Hematol Oncol Clin North Am 10 (2): 293-320, 1996. [PUBMED Abstract]
Pedersen-Bjergaard J, Philip P: Balanced translocations involving chromosome bands 11q23 and 21q22 are highly characteristic of myelodysplasia and leukemia following therapy with cytostatic agents targeting at DNA-topoisomerase II. Blood 78 (4): 1147-8, 1991. [PUBMED Abstract]
Kok JL, Teepen JC, van der Pal HJ, et al.: Incidence of and Risk Factors for Histologically Confirmed Solid Benign Tumors Among Long-term Survivors of Childhood Cancer. JAMA Oncol 5 (5): 671-680, 2019. [PUBMED Abstract]
Inskip PD, Sigurdson AJ, Veiga L, et al.: Radiation-Related New Primary Solid Cancers in the Childhood Cancer Survivor Study: Comparative Radiation Dose Response and Modification of Treatment Effects. Int J Radiat Oncol Biol Phys 94 (4): 800-7, 2016. [PUBMED Abstract]
Ehrhardt MJ, Howell CR, Hale K, et al.: Subsequent Breast Cancer in Female Childhood Cancer Survivors in the St Jude Lifetime Cohort Study (SJLIFE). J Clin Oncol 37 (19): 1647-1656, 2019. [PUBMED Abstract]
Taylor AJ, Little MP, Winter DL, et al.: Population-based risks of CNS tumors in survivors of childhood cancer: the British Childhood Cancer Survivor Study. J Clin Oncol 28 (36): 5287-93, 2010. [PUBMED Abstract]
Henderson TO, Whitton J, Stovall M, et al.: Secondary sarcomas in childhood cancer survivors: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 99 (4): 300-8, 2007. [PUBMED Abstract]
Veiga LH, Holmberg E, Anderson H, et al.: Thyroid Cancer after Childhood Exposure to External Radiation: An Updated Pooled Analysis of 12 Studies. Radiat Res 185 (5): 473-84, 2016. [PUBMED Abstract]
Reulen RC, Frobisher C, Winter DL, et al.: Long-term risks of subsequent primary neoplasms among survivors of childhood cancer. JAMA 305 (22): 2311-9, 2011. [PUBMED Abstract]
Teepen JC, Kok JL, Kremer LC, et al.: Long-Term Risk of Skin Cancer Among Childhood Cancer Survivors: A DCOG-LATER Cohort Study. J Natl Cancer Inst 111 (8): 845-853, 2019. [PUBMED Abstract]
Majhail NS, Brazauskas R, Rizzo JD, et al.: Secondary solid cancers after allogeneic hematopoietic cell transplantation using busulfan-cyclophosphamide conditioning. Blood 117 (1): 316-22, 2011. [PUBMED Abstract]
Baker KS, Leisenring WM, Goodman PJ, et al.: Total body irradiation dose and risk of subsequent neoplasms following allogeneic hematopoietic cell transplantation. Blood 133 (26): 2790-2799, 2019. [PUBMED Abstract]
Veiga LH, Curtis RE, Morton LM, et al.: Association of Breast Cancer Risk After Childhood Cancer With Radiation Dose to the Breast and Anthracycline Use: A Report From the Childhood Cancer Survivor Study. JAMA Pediatr 173 (12): 1171-1179, 2019. [PUBMED Abstract]
Kenney LB, Yasui Y, Inskip PD, et al.: Breast cancer after childhood cancer: a report from the Childhood Cancer Survivor Study. Ann Intern Med 141 (8): 590-7, 2004. [PUBMED Abstract]
Moskowitz CS, Chou JF, Wolden SL, et al.: Breast cancer after chest radiation therapy for childhood cancer. J Clin Oncol 32 (21): 2217-23, 2014. [PUBMED Abstract]
Henderson TO, Liu Q, Turcotte LM, et al.: Association of Changes in Cancer Therapy Over 3 Decades With Risk of Subsequent Breast Cancer Among Female Childhood Cancer Survivors: A Report From the Childhood Cancer Survivor Study (CCSS). JAMA Oncol : , 2022. [PUBMED Abstract]
Schaapveld M, Aleman BM, van Eggermond AM, et al.: Second Cancer Risk Up to 40 Years after Treatment for Hodgkin’s Lymphoma. N Engl J Med 373 (26): 2499-511, 2015. [PUBMED Abstract]
Travis LB, Hill D, Dores GM, et al.: Cumulative absolute breast cancer risk for young women treated for Hodgkin lymphoma. J Natl Cancer Inst 97 (19): 1428-37, 2005. [PUBMED Abstract]
O’Brien MM, Donaldson SS, Balise RR, et al.: Second malignant neoplasms in survivors of pediatric Hodgkin’s lymphoma treated with low-dose radiation and chemotherapy. J Clin Oncol 28 (7): 1232-9, 2010. [PUBMED Abstract]
Inskip PD, Robison LL, Stovall M, et al.: Radiation dose and breast cancer risk in the childhood cancer survivor study. J Clin Oncol 27 (24): 3901-7, 2009. [PUBMED Abstract]
Travis LB, Hill DA, Dores GM, et al.: Breast cancer following radiotherapy and chemotherapy among young women with Hodgkin disease. JAMA 290 (4): 465-75, 2003. [PUBMED Abstract]
van Leeuwen FE, Klokman WJ, Stovall M, et al.: Roles of radiation dose, chemotherapy, and hormonal factors in breast cancer following Hodgkin’s disease. J Natl Cancer Inst 95 (13): 971-80, 2003. [PUBMED Abstract]
Lange JM, Takashima JR, Peterson SM, et al.: Breast cancer in female survivors of Wilms tumor: a report from the national Wilms tumor late effects study. Cancer 120 (23): 3722-30, 2014. [PUBMED Abstract]
Wang Y, Ronckers CM, van Leeuwen FE, et al.: Subsequent female breast cancer risk associated with anthracycline chemotherapy for childhood cancer. Nat Med 29 (9): 2268-2277, 2023. [PUBMED Abstract]
Henderson TO, Moskowitz CS, Chou JF, et al.: Breast Cancer Risk in Childhood Cancer Survivors Without a History of Chest Radiotherapy: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 34 (9): 910-8, 2016. [PUBMED Abstract]
Eisenberg ER, Weiss A, Prakash I, et al.: Surgical Management and Contralateral Breast Cancer Risk in Women with History of Radiation Therapy for Hodgkin Lymphoma: Results from a Population-Based Cohort. Ann Surg Oncol 29 (11): 6673-6680, 2022. [PUBMED Abstract]
Moskowitz CS, Ronckers CM, Chou JF, et al.: Development and Validation of a Breast Cancer Risk Prediction Model for Childhood Cancer Survivors Treated With Chest Radiation: A Report From the Childhood Cancer Survivor Study and the Dutch Hodgkin Late Effects and LATER Cohorts. J Clin Oncol 39 (27): 3012-3021, 2021. [PUBMED Abstract]
Dores GM, Anderson WF, Beane Freeman LE, et al.: Risk of breast cancer according to clinicopathologic features among long-term survivors of Hodgkin’s lymphoma treated with radiotherapy. Br J Cancer 103 (7): 1081-4, 2010. [PUBMED Abstract]
Horst KC, Hancock SL, Ognibene G, et al.: Histologic subtypes of breast cancer following radiotherapy for Hodgkin lymphoma. Ann Oncol 25 (4): 848-51, 2014. [PUBMED Abstract]
Demoor-Goldschmidt C, Supiot S, Mahé MA, et al.: Clinical and histological features of second breast cancers following radiotherapy for childhood and young adult malignancy. Br J Radiol 91 (1086): 20170824, 2018. [PUBMED Abstract]
Castiglioni F, Terenziani M, Carcangiu ML, et al.: Radiation effects on development of HER2-positive breast carcinomas. Clin Cancer Res 13 (1): 46-51, 2007. [PUBMED Abstract]
Gaffney DK, Hemmersmeier J, Holden J, et al.: Breast cancer after mantle irradiation for Hodgkin’s disease: correlation of clinical, pathologic, and molecular features including loss of heterozygosity at BRCA1 and BRCA2. Int J Radiat Oncol Biol Phys 49 (2): 539-46, 2001. [PUBMED Abstract]
Wong SM, Ajjamada L, Weiss AC, et al.: Clinicopathologic features of breast cancers diagnosed in women treated with prior radiation therapy for Hodgkin lymphoma: Results from a population-based cohort. Cancer 128 (7): 1365-1372, 2022. [PUBMED Abstract]
Moskowitz CS, Chou JF, Neglia JP, et al.: Mortality After Breast Cancer Among Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 37 (24): 2120-2130, 2019. [PUBMED Abstract]
Yeh JM, Lowry KP, Schechter CB, et al.: Clinical Benefits, Harms, and Cost-Effectiveness of Breast Cancer Screening for Survivors of Childhood Cancer Treated With Chest Radiation : A Comparative Modeling Study. Ann Intern Med 173 (5): 331-341, 2020. [PUBMED Abstract]
Vivanco M, Dalle JH, Alberti C, et al.: Malignant and benign thyroid nodules after total body irradiation preceding hematopoietic cell transplantation during childhood. Eur J Endocrinol 167 (2): 225-33, 2012. [PUBMED Abstract]
Friedman DN, Goodman PJ, Leisenring WM, et al.: Impact of risk-based therapy on late morbidity and mortality in neuroblastoma survivors: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 116 (6): 885-894, 2024. [PUBMED Abstract]
Michaelson EM, Chen YH, Silver B, et al.: Thyroid malignancies in survivors of Hodgkin lymphoma. Int J Radiat Oncol Biol Phys 88 (3): 636-41, 2014. [PUBMED Abstract]
Sigurdson AJ, Ronckers CM, Mertens AC, et al.: Primary thyroid cancer after a first tumour in childhood (the Childhood Cancer Survivor Study): a nested case-control study. Lancet 365 (9476): 2014-23, 2005 Jun 11-17. [PUBMED Abstract]
Clement SC, Lebbink CA, Klein Hesselink MS, et al.: Presentation and outcome of subsequent thyroid cancer among childhood cancer survivors compared to sporadic thyroid cancer: a matched national study. Eur J Endocrinol 183 (2): 169-180, 2020. [PUBMED Abstract]
Casey DL, Vogelius IR, Brodin NP, et al.: Risk of Subsequent Neoplasms in Childhood Cancer Survivors After Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 640-654, 2024. [PUBMED Abstract]
Hijiya N, Hudson MM, Lensing S, et al.: Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA 297 (11): 1207-15, 2007. [PUBMED Abstract]
Neglia JP, Robison LL, Stovall M, et al.: New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 98 (21): 1528-37, 2006. [PUBMED Abstract]
Faraci M, Morana G, Bagnasco F, et al.: Magnetic resonance imaging in childhood leukemia survivors treated with cranial radiotherapy: a cross sectional, single center study. Pediatr Blood Cancer 57 (2): 240-6, 2011. [PUBMED Abstract]
Vinchon M, Leblond P, Caron S, et al.: Radiation-induced tumors in children irradiated for brain tumor: a longitudinal study. Childs Nerv Syst 27 (3): 445-53, 2011. [PUBMED Abstract]
Koike T, Yanagimachi N, Ishiguro H, et al.: High incidence of radiation-induced cavernous hemangioma in long-term survivors who underwent hematopoietic stem cell transplantation with radiation therapy during childhood or adolescence. Biol Blood Marrow Transplant 18 (7): 1090-8, 2012. [PUBMED Abstract]
Heymer EJ, Hawkins MM, Winter DL, et al.: Risk of subsequent gliomas and meningiomas among 69,460 5-year survivors of childhood and adolescent cancer in Europe: the PanCareSurFup study. Br J Cancer 130 (6): 976-986, 2024. [PUBMED Abstract]
Bowers DC, Verbruggen LC, Kremer LCM, et al.: Surveillance for subsequent neoplasms of the CNS for childhood, adolescent, and young adult cancer survivors: a systematic review and recommendations from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol 22 (5): e196-e206, 2021. [PUBMED Abstract]
Baliga S, Gallotto S, Bajaj B, et al.: Decade-long disease, secondary malignancy, and brainstem injury outcomes in pediatric and young adult medulloblastoma patients treated with proton radiotherapy. Neuro Oncol 24 (6): 1010-1019, 2022. [PUBMED Abstract]
Wong JR, Morton LM, Tucker MA, et al.: Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol 32 (29): 3284-90, 2014. [PUBMED Abstract]
Fidler MM, Reulen RC, Winter DL, et al.: Risk of Subsequent Bone Cancers Among 69 460 Five-Year Survivors of Childhood and Adolescent Cancer in Europe. J Natl Cancer Inst 110 (2): , 2018. [PUBMED Abstract]
Bright CJ, Hawkins MM, Winter DL, et al.: Risk of Soft-Tissue Sarcoma Among 69 460 Five-Year Survivors of Childhood Cancer in Europe. J Natl Cancer Inst 110 (6): 649-660, 2018. [PUBMED Abstract]
Chaussade A, Millot G, Wells C, et al.: Correlation between RB1germline mutations and second primary malignancies in hereditary retinoblastoma patients treated with external beam radiotherapy. Eur J Med Genet 62 (3): 217-223, 2019. [PUBMED Abstract]
Kube SJ, Blattmann C, Bielack SS, et al.: Secondary malignant neoplasms after bone and soft tissue sarcomas in children, adolescents, and young adults. Cancer 128 (9): 1787-1800, 2022. [PUBMED Abstract]
Schwartz B, Benadjaoud MA, Cléro E, et al.: Risk of second bone sarcoma following childhood cancer: role of radiation therapy treatment. Radiat Environ Biophys 53 (2): 381-90, 2014. [PUBMED Abstract]
Bielack SS, Blattmann C, Hassenpflug W, et al.: Osteosarcoma Arising After Ewing Sarcoma or Vice Versa: A Report of 20 Affected Patients from the Cooperative Osteosarcoma Study Group (COSS). Anticancer Res 43 (11): 4975-4981, 2023. [PUBMED Abstract]
Henderson TO, Rajaraman P, Stovall M, et al.: Risk factors associated with secondary sarcomas in childhood cancer survivors: a report from the childhood cancer survivor study. Int J Radiat Oncol Biol Phys 84 (1): 224-30, 2012. [PUBMED Abstract]
Reulen RC, Winter DL, Diallo I, et al.: Risk Factors for Primary Bone Cancer After Childhood Cancer: A PanCare Childhood and Adolescent Cancer Survivor Care and Follow-Up Studies Nested Case-Control Study. J Clin Oncol 41 (21): 3735-3746, 2023. [PUBMED Abstract]
MacCarthy A, Bayne AM, Brownbill PA, et al.: Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951-2004. Br J Cancer 108 (12): 2455-63, 2013. [PUBMED Abstract]
Temming P, Arendt M, Viehmann A, et al.: Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr Blood Cancer 64 (1): 71-80, 2017. [PUBMED Abstract]
Kleinerman RA, Schonfeld SJ, Sigel BS, et al.: Bone and Soft-Tissue Sarcoma Risk in Long-Term Survivors of Hereditary Retinoblastoma Treated With Radiation. J Clin Oncol 37 (35): 3436-3445, 2019. [PUBMED Abstract]
Thorsness SL, Freites-Martinez A, Marchetti MA, et al.: Nonmelanoma Skin Cancer in Childhood and Young Adult Cancer Survivors Previously Treated With Radiotherapy. J Natl Compr Canc Netw 17 (3): 237-243, 2019. [PUBMED Abstract]
Boull C, Chen Y, Im C, et al.: Keratinocyte carcinomas in survivors of childhood cancer: A report from the childhood cancer survivor study. J Am Acad Dermatol 91 (6): 1125-1135, 2024. [PUBMED Abstract]
Geller AC, Coroiu A, Keske RR, et al.: Advancing Survivors Knowledge (ASK Study) of Skin Cancer Surveillance After Childhood Cancer: A Randomized Controlled Trial in the Childhood Cancer Survivor Study. J Clin Oncol 41 (12): 2269-2280, 2023. [PUBMED Abstract]
Rotz SJ, Stratton K, Leisenring WM, et al.: Melanoma Among Adult Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 43 (10): 1219-1228, 2025. [PUBMED Abstract]
Braam KI, Overbeek A, Kaspers GJ, et al.: Malignant melanoma as second malignant neoplasm in long-term childhood cancer survivors: a systematic review. Pediatr Blood Cancer 58 (5): 665-74, 2012. [PUBMED Abstract]
Kleinerman RA, Schonfeld SJ, Abramson DH, et al.: Increased Risk of Skin Cancer in 1,851 Long-Term Retinoblastoma Survivors. J Invest Dermatol 141 (12): 2849-2857.e3, 2021. [PUBMED Abstract]
Ghosh T, Chen Y, Dietz AC, et al.: Lung Cancer as a Subsequent Malignant Neoplasm in Survivors of Childhood Cancer. Cancer Epidemiol Biomarkers Prev 30 (12): 2235-2243, 2021. [PUBMED Abstract]
van Leeuwen FE, Klokman WJ, Stovall M, et al.: Roles of radiotherapy and smoking in lung cancer following Hodgkin’s disease. J Natl Cancer Inst 87 (20): 1530-7, 1995. [PUBMED Abstract]
Henderson TO, Oeffinger KC, Whitton J, et al.: Secondary gastrointestinal cancer in childhood cancer survivors: a cohort study. Ann Intern Med 156 (11): 757-66, W-260, 2012. [PUBMED Abstract]
Heymer EJ, Jóźwiak K, Kremer LC, et al.: Cumulative Absolute Risk of Subsequent Colorectal Cancer After Abdominopelvic Radiotherapy Among Childhood Cancer Survivors: A PanCareSurFup Study. J Clin Oncol 42 (3): 336-347, 2024. [PUBMED Abstract]
Tukenova M, Diallo I, Anderson H, et al.: Second malignant neoplasms in digestive organs after childhood cancer: a cohort-nested case-control study. Int J Radiat Oncol Biol Phys 82 (3): e383-90, 2012. [PUBMED Abstract]
Daly PE, Samiee S, Cino M, et al.: High prevalence of adenomatous colorectal polyps in young cancer survivors treated with abdominal radiation therapy: results of a prospective trial. Gut 66 (10): 1797-1801, 2017. [PUBMED Abstract]
Teepen JC, Kok JL, van Leeuwen FE, et al.: Colorectal Adenomas and Cancers After Childhood Cancer Treatment: A DCOG-LATER Record Linkage Study. J Natl Cancer Inst 110 (7): 758-767, 2018. [PUBMED Abstract]
Reulen RC, Wong KF, Bright CJ, et al.: Risk of digestive cancers in a cohort of 69 460 five-year survivors of childhood cancer in Europe: the PanCareSurFup study. Gut : , 2020. [PUBMED Abstract]
Geurts YM, Shakir R, Ntentas G, et al.: Association of Radiation and Procarbazine Dose With Risk of Colorectal Cancer Among Survivors of Hodgkin Lymphoma. JAMA Oncol 9 (4): 481-489, 2023. [PUBMED Abstract]
Sunguc C, Hawkins MM, Winter DL, et al.: Risk of subsequent primary oral cancer in a cohort of 69,460 5-year survivors of childhood and adolescent cancer in Europe: the PanCareSurFup study. Br J Cancer 128 (1): 80-90, 2023. [PUBMED Abstract]
Liu JJ, De Vivo I, Wu CY, et al.: Subsequent primary urogenital cancers among childhood and adolescent cancer survivors in the United States. Urol Oncol 40 (2): 65.e11-65.e18, 2022. [PUBMED Abstract]
Wilson CL, Ness KK, Neglia JP, et al.: Renal carcinoma after childhood cancer: a report from the childhood cancer survivor study. J Natl Cancer Inst 105 (7): 504-8, 2013. [PUBMED Abstract]
de Vathaire F, Scwhartz B, El-Fayech C, et al.: Risk of a Second Kidney Carcinoma Following Childhood Cancer: Role of Chemotherapy and Radiation Dose to Kidneys. J Urol 194 (5): 1390-5, 2015. [PUBMED Abstract]
Hedgepeth RC, Zhou M, Ross J: Rapid development of metastatic Xp11 translocation renal cell carcinoma in a girl treated for neuroblastoma. J Pediatr Hematol Oncol 31 (8): 602-4, 2009. [PUBMED Abstract]
Argani P, Laé M, Ballard ET, et al.: Translocation carcinomas of the kidney after chemotherapy in childhood. J Clin Oncol 24 (10): 1529-34, 2006. [PUBMED Abstract]
Ma J, Pan C, Yin M: Translocation Renal Cell Carcinoma in a Child Previously Treated for Infantile Fibrosarcoma. Pediatr Dev Pathol 21 (4): 418-422, 2018 Jul-Aug. [PUBMED Abstract]
Fleitz JM, Wootton-Gorges SL, Wyatt-Ashmead J, et al.: Renal cell carcinoma in long-term survivors of advanced stage neuroblastoma in early childhood. Pediatr Radiol 33 (8): 540-5, 2003. [PUBMED Abstract]
Henderson TO, Fowler BW, Hamann HA, et al.: Subsequent malignant neoplasms in the Childhood Cancer Survivor Study: Occurrence of cancer types in which human papillomavirus is an established etiologic risk factor. Cancer 128 (2): 373-382, 2022. [PUBMED Abstract]
Ou JY, Bennion N, Parker K, et al.: Risk Factors and Trends for HPV-Associated Subsequent Malignant Neoplasms among Adolescent and Young Adult Cancer Survivors. Cancer Epidemiol Biomarkers Prev 32 (5): 625-633, 2023. [PUBMED Abstract]
Brown AL, Arroyo VM, Agrusa JE, et al.: Survival disparities for second primary malignancies diagnosed among childhood cancer survivors: A population-based assessment. Cancer 125 (20): 3623-3630, 2019. [PUBMED Abstract]
Archer NM, Amorim RP, Naves R, et al.: An Increased Risk of Second Malignant Neoplasms After Rhabdomyosarcoma: Population-Based Evidence for a Cancer Predisposition Syndrome? Pediatr Blood Cancer 63 (2): 196-201, 2016. [PUBMED Abstract]
Wang X, Sun CL, Hageman L, et al.: Clinical and Genetic Risk Prediction of Subsequent CNS Tumors in Survivors of Childhood Cancer: A Report From the COG ALTE03N1 Study. J Clin Oncol 35 (32): 3688-3696, 2017. [PUBMED Abstract]
Waespe N, Belle FN, Redmond S, et al.: Cancer predisposition syndromes as a risk factor for early second primary neoplasms after childhood cancer – A national cohort study. Eur J Cancer 145: 71-80, 2021. [PUBMED Abstract]
Andersson A, Enblad G, Tavelin B, et al.: Family history of cancer as a risk factor for second malignancies after Hodgkin’s lymphoma. Br J Cancer 98 (5): 1001-5, 2008. [PUBMED Abstract]
Hisada M, Garber JE, Fung CY, et al.: Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst 90 (8): 606-11, 1998. [PUBMED Abstract]
de Andrade KC, Khincha PP, Hatton JN, et al.: Cancer incidence, patterns, and genotype-phenotype associations in individuals with pathogenic or likely pathogenic germline TP53 variants: an observational cohort study. Lancet Oncol 22 (12): 1787-1798, 2021. [PUBMED Abstract]
Bhatia S, Chen Y, Wong FL, et al.: Subsequent Neoplasms After a Primary Tumor in Individuals With Neurofibromatosis Type 1. J Clin Oncol 37 (32): 3050-3058, 2019. [PUBMED Abstract]
de Blank P, Li N, Fisher MJ, et al.: Late morbidity and mortality in adult survivors of childhood glioma with neurofibromatosis type 1: report from the Childhood Cancer Survivor Study. Genet Med 22 (11): 1794-1802, 2020. [PUBMED Abstract]
Strahm B, Malkin D: Hereditary cancer predisposition in children: genetic basis and clinical implications. Int J Cancer 119 (9): 2001-6, 2006. [PUBMED Abstract]
Cullinan N, Schiller I, Di Giuseppe G, et al.: Utility of a Cancer Predisposition Screening Tool for Predicting Subsequent Malignant Neoplasms in Childhood Cancer Survivors. J Clin Oncol 39 (29): 3207-3216, 2021. [PUBMED Abstract]
Goudie C, Witkowski L, Cullinan N, et al.: Performance of the McGill Interactive Pediatric OncoGenetic Guidelines for Identifying Cancer Predisposition Syndromes. JAMA Oncol 7 (12): 1806-1814, 2021. [PUBMED Abstract]
Qin N, Wang Z, Liu Q, et al.: Pathogenic Germline Mutations in DNA Repair Genes in Combination With Cancer Treatment Exposures and Risk of Subsequent Neoplasms Among Long-Term Survivors of Childhood Cancer. J Clin Oncol 38 (24): 2728-2740, 2020. [PUBMED Abstract]
Collins A, Harrington V: Repair of oxidative DNA damage: assessing its contribution to cancer prevention. Mutagenesis 17 (6): 489-93, 2002. [PUBMED Abstract]
Gibson TM, Karyadi DM, Hartley SW, et al.: Polygenic risk scores, radiation treatment exposures and subsequent cancer risk in childhood cancer survivors. Nat Med 30 (3): 690-698, 2024. [PUBMED Abstract]
Im C, Sharafeldin N, Yuan Y, et al.: Polygenic Risk and Chemotherapy-Related Subsequent Malignancies in Childhood Cancer Survivors: A Childhood Cancer Survivor Study and St Jude Lifetime Cohort Study Report. J Clin Oncol 41 (27): 4381-4393, 2023. [PUBMED Abstract]
Landier W, Bhatia S, Eshelman DA, et al.: Development of risk-based guidelines for pediatric cancer survivors: the Children’s Oncology Group Long-Term Follow-Up Guidelines from the Children’s Oncology Group Late Effects Committee and Nursing Discipline. J Clin Oncol 22 (24): 4979-90, 2004. [PUBMED Abstract]
Kremer LC, Mulder RL, Oeffinger KC, et al.: A worldwide collaboration to harmonize guidelines for the long-term follow-up of childhood and young adult cancer survivors: a report from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Pediatr Blood Cancer 60 (4): 543-9, 2013. [PUBMED Abstract]
Mulder RL, Hudson MM, Bhatia S, et al.: Updated Breast Cancer Surveillance Recommendations for Female Survivors of Childhood, Adolescent, and Young Adult Cancer From the International Guideline Harmonization Group. J Clin Oncol 38 (35): 4194-4207, 2020. [PUBMED Abstract]
Kriege M, Brekelmans CT, Boetes C, et al.: Efficacy of MRI and mammography for breast-cancer screening in women with a familial or genetic predisposition. N Engl J Med 351 (5): 427-37, 2004. [PUBMED Abstract]
Leach MO, Boggis CR, Dixon AK, et al.: Screening with magnetic resonance imaging and mammography of a UK population at high familial risk of breast cancer: a prospective multicentre cohort study (MARIBS). Lancet 365 (9473): 1769-78, 2005 May 21-27. [PUBMED Abstract]
Tieu MT, Cigsar C, Ahmed S, et al.: Breast cancer detection among young survivors of pediatric Hodgkin lymphoma with screening magnetic resonance imaging. Cancer 120 (16): 2507-13, 2014. [PUBMED Abstract]
Saslow D, Boetes C, Burke W, et al.: American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 57 (2): 75-89, 2007 Mar-Apr. [PUBMED Abstract]
Berrington de Gonzalez A, Berg CD, Visvanathan K, et al.: Estimated risk of radiation-induced breast cancer from mammographic screening for young BRCA mutation carriers. J Natl Cancer Inst 101 (3): 205-9, 2009. [PUBMED Abstract]
Young KC, Burch A, Oduko JM: Radiation doses received in the UK Breast Screening Programme in 2001 and 2002. Br J Radiol 78 (927): 207-18, 2005. [PUBMED Abstract]
Spelic DC: Trends in Mammography Dose and Image Quality 1974-2005. Silver Spring, Md: U.S. Food and Drug Administration, 2006. Available online. Last accessed August 21, 2023.
Clement SC, Kremer LCM, Verburg FA, et al.: Balancing the benefits and harms of thyroid cancer surveillance in survivors of Childhood, adolescent and young adult cancer: Recommendations from the international Late Effects of Childhood Cancer Guideline Harmonization Group in collaboration with the PanCareSurFup Consortium. Cancer Treat Rev 63: 28-39, 2018. [PUBMED Abstract]
Late Effects of the Cardiovascular System
Cardiovascular disease, after recurrence of the original cancer and development of second primary cancers, has been reported to be the leading cause of premature mortality among long-term childhood cancer survivors.[1–3]
Evidence (excess risk of premature cardiovascular mortality):
In the North American Childhood Cancer Survivor Study (CCSS), more than 34,000 5-year survivors of childhood cancer were diagnosed and treated from 1970 to 1999.[1]
Participants followed for 15 years had a standardized mortality ratio (SMR) of 11.7 (95% confidence interval [CI], 9.4–14.4) for cardiac mortality.
Late cardiac mortality in children who were treated more recently (i.e., in the 1990s) appears to have decreased (e.g., the cumulative incidence was 0.5% in 1970–1974 vs. 0.1% in 1990–1994).
In the CCSS, outcomes for patients diagnosed and treated in adolescence and early adulthood (aged >15 to <21 years) were compared with survivors of similar cancers diagnosed in childhood (aged <15 years) and the general population.[4]
Childhood cancer survivors (n = 5,804) had a greater SMR than early-adolescent and young adult survivors (n = 5,804) for cardiac deaths beginning 20 years after diagnosis (SMR, 5.4; 95% CI, 3.7–7.8 vs. SMR, 1.8; 95% CI, 0.7–4.3).
When compared with siblings of the same age, survivors of childhood cancer (n = 4,082) had a higher risk of developing grades 3 to 5 chronic cardiac conditions than early adolescents and young adults (n = 4,082) (hazard ratio [HR], 5.6 [4.5–7.1] vs. 4.3 [3.5–5.4]).
The incidence and accumulation of nonmajor adverse cardiovascular events, including subclinical conditions found by screening, increases the risk of subsequent major adverse cardiovascular events in survivors of childhood cancer. A prospective longitudinal cohort study that used systematic in-person clinical assessments (St. Jude Lifetime [SJLIFE] cohort consisting of 9,602 5-year survivors and 737 community controls) reported the following:[5]
Increasing cumulative burden of grades 1 to 4 nonmajor adverse cardiovascular events was associated with an increased future risk of major adverse cardiovascular events (one condition: relative risk [RR], 4.3; P < .0001; two conditions: RR, 6.6; P < .0001; and three conditions: RR, 7.7; P < .0001).
Increased risk of major adverse cardiovascular events was observed with specific subclinical conditions such as grade 1 arrythmias (RR, 1.5; P = .0017), grade 2 left ventricular systolic dysfunction (RR, 2.2; P < .0001), and grade 2 valvular disorders (RR, 2.2; P = .013).
Grade 1 hypercholesterolemia, grades 1 to 2 hypertriglyceridemia, or grades 1 to 2 vascular stenosis did not increase the risk of major adverse cardiovascular events.
Cardiac disease becomes increasingly important as survivors of childhood cancer reach mature adulthood. This finding was observed in the population-based British Childhood Cancer Survivor Study, comprised of 34,489 5-year survivors of childhood cancer diagnosed from 1940 to 2006.[2,6]
In survivors of childhood cancer aged 60 years and older, circulatory disease overtakes subsequent primary neoplasms as the leading cause of excess mortality (37% of the excess number of deaths observed were caused by circulatory conditions compared with 31% of excess number of deaths caused by subsequent primary neoplasms).[2]
The risk of both overall cardiac and cardiomyopathy/heart failure mortality was greatest among those diagnosed between 1980 and 1989. Survivors who were diagnosed from 1980 to 1989 had 28.9 times the excess number of cardiac deaths as did survivors who were diagnosed either before 1970 or from 1990 to the present.[6]
The specific late effects covered in this section include the following:
Cardiomyopathy/heart failure.
Ischemic heart disease.
Pericardial heart disease.
Valve disease.
Conduction disorders.
Cerebrovascular disease.
Venous thromboembolism.
This section will also briefly discuss the influence of related conditions such as hypertension, dyslipidemia, and diabetes. However, this section will not provide a detailed review of those conditions as a consequence of childhood cancer treatment. A comprehensive review of long-term cardiovascular toxicity in childhood and young adult survivors of cancer has been published.[7]
Cardiovascular Outcomes
Numerous studies focus on cardiac events among childhood cancer survivors. Very large cohort studies exist, many with several decades of follow-up, that are either hospital based,[8–10] clinical trial based,[11,12] or population based.[2,3,6]
Notably, the average age of these populations is still relatively young (early or mid-adulthood). Consequently, the reported risk of serious cardiovascular outcomes is very high relative to the age-matched general population, whereas the absolute risk often remains low, limiting the power of many studies.
Among the large studies featuring thousands of survivors, the main limitation has been inadequate ability to clinically ascertain late cardiovascular complications, with a greater reliance on either administrative records (e.g., death registries) and/or self-report or proxy-report.
While each study design has some inherent biases, the overall literature, based on a combination of self-reported outcomes, clinical ascertainment, and administrative data sources, is robust. These studies conclude that certain cancer-related exposures are associated with a significantly greater risk of cardiovascular morbidity and mortality in survivors.
Although late effects research often lags behind changes in contemporary therapy, many therapies linked to cardiovascular late effects remain in common use today. However, restricted doses of these therapies are often given to patients with biologically favorable disease presentations. In addition, dexrazoxane cardioprotection is also widely used for patients exposed to these therapies.[13–15]
Ongoing research is important to ensure that use of newer targeted agents does not result in unexpected cardiovascular effects.[16]
A case-control study of the PanCareSurFup and ProCardio cohort evaluated treatment-related risk factors for heart failure in survivors of childhood cancer (5 or more years) diagnosed between 1940 and 2009.[17]
The 50-year cumulative incidence of heart failure among the entire cohort was 2% (95% CI, 1.7%–2.2%).
A subcohort study evaluated 500 cases with symptomatic heart failure and 500 controls without heart failure (matched for sex, age, calendar year of first diagnosis, and length of follow-up) for whom detailed treatment exposures were available. The risk of heart failure was increased more than fivefold (odds ratio [OR], 5.5; 95% CI, 2.5–12.3) among survivors who received a mean heart radiation therapy dose of 5 Gy to 15 Gy or less, compared with those who did not receive heart radiation therapy. The risk increased with a larger volume of radiation exposing the heart.
Heart failure risk increased in a linear fashion with higher mean heart radiation therapy doses.
Survivors who received a cumulative anthracycline dose of more than 100 mg/m2 had a substantially increased risk of heart failure (OR, 5.8; 95% CI, 2.9–11.3 for 100 to <250 mg/m2 and OR, 21.2; 95% CI, 11.4–39.2 for >250 mg/m2).
An anthracycline dose of less than 100 mg/m2 was not a significant risk factor for heart failure.
CCSS investigators reported on major cardiac events among participants diagnosed with childhood cancer between 1970 and 1999.[18]
In this update, the 20-year cumulative incidence of heart failure and coronary artery disease for patients treated in the 1990s declined over the decades to 0.54% and 0.19%, respectively, but was significant only for coronary artery disease.
The risk of coronary artery disease was significantly decreased from the 1970s, 1980s, and 1990s (0.38%, 0.24%, and 0.19%, respectively; HR, 0.65) and was attributed to historical reductions in exposure to cardiac radiation, particularly among survivors of Hodgkin lymphoma.
For patients treated in the 1990s, the 20-year cumulative incidence was 0.05% for valvular disease, 0.03% for pericardial disease, and 0.13% for arrhythmias. These numbers did not change over the eras (1970–1990).
In the CCSS, data from 24,214 5-year survivors diagnosed between 1970 and 1999 were used to assess the impacts of radiation therapy dose and exposed cardiac volume, select chemotherapeutic agents, and age at exposure on the risk of late-onset cardiac disease.[19]
The cumulative incidence of cardiac disease (any cardiac disease, coronary artery disease, and heart failure) 30 years from diagnosis was 4.8%. Male survivors were more likely to develop coronary artery disease and less likely to develop heart failure than were female survivors. Non-Hispanic Black survivors were more likely to develop any cardiac disease than were non-Hispanic White survivors.
Low-to-moderate radiation therapy doses (5.0–19.9 Gy) to large cardiac volumes (>50% of the heart) were associated with a 1.6-fold increased risk of cardiac disease compared with survivors who did not have any cardiac radiation therapy exposure.
High doses (>20 Gy) to small cardiac volumes (0.1%–29.9%) were associated with an elevated rate of cardiac disease compared with unexposed survivors.
A dose-response relationship was observed between anthracycline exposure and heart failure, with younger children (<13 years) at the greatest risk of heart failure after comparable dosing.
Another CCSS study evaluated radiation dose–response relationships for cardiac substructures and cardiac outcomes among 25,481 5-year survivors treated between 1970 to 1999. Of these survivors, 48.2% were exposed to radiation therapy.[20]
At a median age of 30 years, the cumulative incidence (35 years from diagnosis) was 3.9% (95% CI, 3.4%–4.3%) for coronary artery disease, 3.8% (95% CI, 3.4%–4.2%) for heart failure, 1.2% (95% CI, 1.0%–1.5%) for valvular disease, and 1.4% (95% CI, 1.1%–1.96%) for arrythmia.
Mean doses of 5 to 9.9 Gy to the whole heart did not increase the risk of any cardiac disease, whereas mean doses of 5 to 9.9 Gy to the right coronary artery (RR, 2.6; 95% CI, 1.6–4.1) and left ventricle (RR, 2.2; 95% CI, 1.3–3.7) increased the risk of coronary artery disease.
Mean doses of 5 to 9.9 Gy to the tricuspid valve (RR, 5.5; 95% CI, 2.0–15.1) and right ventricle (RR, 8.4; 95% CI, 3.7–19.0) increased the risk of valvular disease.
Statistical modeling suggested that any radiation to the cardiac substructures elevate the risk of cardiac diseases, and there is no threshold dose that can mitigate this risk.
A cross-sectional CCSS study included 571 adult-aged survivors of childhood cancer (median age, 37.7 years; 28.5 years from cancer diagnosis). The study compared rates of underdiagnosis and undertreatment of modifiable risk factors among survivors and respondents to the National Health and Nutrition Examination Survey (NHNES).[21]
Rates of cardiovascular disease risk factor underdiagnosis were similar (27.1% of survivors vs. 26.1% of participants from the NHNES; P = .73). However, survivors were more likely to be undertreated (21.0% versus 13.9%, P = .007; OR, 1.8; 95% CI, 1.2–2.7).
Hypertension (18.9%) and dyslipidemia (16.3%) represented the most underdiagnosed and undertreated risk factors.
Men and survivors who were overweight or had obesity were more likely to be underdiagnosed and undertreated.
These adult-aged childhood cancer survivors were nearly twice as likely to be undertreated for these conditions.
Dyslipidemia is more common and is an independent predictor of cardiovascular disease among survivors, compared with controls. Specific lipid abnormalities and the risk of atherosclerotic cardiovascular disease were investigated in 4,115 childhood cancer survivors from the SJLIFE cohort.[22]
Of this cohort, 3,406 survivors did not have a previous dyslipidemia diagnosis.
Low HDL cholesterol (HR, 2.9) and elevated triglycerides (HR, 3.1) were associated with an increased risk of myocardial infarction.
Diagnosis of high LDL cholesterol (HR, 2.2), high non-HDL cholesterol (HR, 2.2), low HDL cholesterol (HR, 3.9), and elevated triglycerides (HR, 3.8) were associated with an increased risk of cardiomyopathy.
A Dutch CCSS-LATER study evaluated the prevalence of and risk factors for hypertension in childhood cancer survivors (median age, 32.5 years; median follow-up, 25.5 years) who were treated with potentially nephrotoxic therapies.[23]
Survivors (16.3%) and controls (18.2%) had comparable rates of hypertension. Undiagnosed hypertension was prevalent in 12% of survivors and 17.8% of controls.
Treatment factors associated with risk of hypertension included abdominal radiation therapy of more than 20 Gy and total-body irradiation (TBI).
Among childhood cancer survivors, glomerular filtration rate (GFR) of less than 60 mL/min/1.73 m2 was significantly associated with hypertension (OR, 3.4; 95% CI, 1.4–8.5).
The CCSS demonstrated that the cumulative incidence of serious cardiac events (myocardial infarction, congestive heart failure, pericardial disease, and valvular abnormalities) in childhood cancer survivors continues to increase beyond age 45 years.[8]
The risk of these events was potentiated (i.e., beyond what would be expected by an additive model) by the presence of concurrent, but potentially modifiable, conditions such as obesity, dyslipidemia, diabetes, and, particularly, hypertension.
Hypertension was independently associated with all serious cardiac outcomes (rate ratios, 6-fold to 19-fold), even after adjustment for anthracycline use and chest irradiation.
Of 670 survivors of Hodgkin lymphoma who were treated at St. Jude Children’s Research Hospital (SJCRH) and survived 10 or more years, 348 patients were clinically assessed in the SJLIFE cohort study.[24]
Overall, survivors had a higher cumulative burden (a novel measurement of disease burden that incorporates multiple health conditions and recurrent events into a single metric) than community controls, with the total grade 3 to 5 cumulative burden among survivors at age 30 years being comparable with that of community controls at 50 years.
At age 50 years, the cumulative incidence of those survivors experiencing at least one grade 3 to grade 5 cardiovascular condition was 45.5% (95% CI, 36.6%–54.3%), compared with 15.7% (95% CI, 7.0%–24.4%) in community controls.
Myocardial infarction and structural heart defects were the major contributors to the excess grade 3 to grade 5 cumulative burden in survivors, whereas there was no notable difference in survivors and community controls at age 50 years for grades 3 to 5 cumulative burden of dyslipidemia and essential hypertension.
Another SJLIFE cohort study compared the prevalence of major and minor electrocardiography (ECG) abnormalities among 2,715 participants and 268 community controls.[25]
Major ECG abnormalities were significantly more prevalent in survivors (10.7%) than in controls (4.9%). The most common abnormalities included isolated ST-T wave abnormalities (7.2%), evidence of myocardial infarction (3.7%), and left ventricular hypertrophy with strain pattern (2.8%).
Treatment exposures predicting increased risk of major abnormalities were anthracycline doses of 300 mg/m2 or greater (OR, 1.7; 95% CI, 1.1–2.5) and cardiac radiation (OR, 2.1; 95% CI, 1.5–2.9 [0.01–19.99 Gy]; OR, 2.6; 95% CI, 1.6–3.9 [20–29.99 Gy]; OR, 10.5; 95% CI, 6.5–16.9 [≥30 Gy]).
Major ECG abnormalities were predictive of all-cause mortality (HR, 4.0; 95% CI, 2.1–7.8).
In the Teenage and Young Adult Cancer Survivor Study, cardiac mortality was investigated in more than 200,000 5-year survivors of adolescent and young adult cancer (aged 15–39 years).[3]
Age at diagnosis and type of cancer were identified as being important in determining risk of cardiac mortality.
The SMRs for all cardiac disease combined was greatest for individuals diagnosed at age 15 to 19 years (4.2), decreasing to 1.2 for individuals aged 35 to 39 years (2-sided P for trend < .0001). This age effect was most apparent for survivors of Hodgkin lymphoma, who were also found to be at greatest risk overall.
Limitations of this study included lack of detailed information on exposures to radiation therapy (doses, fields), exposures to chemotherapy (primarily anthracycline dose), and cardiovascular risk factors (e.g., smoking, obesity, hypertension, diabetes, family history).
Dutch investigators evaluated the risk of heart failure, temporal changes by treatment periods, and the risk factors for heart failure in 6,165 childhood cancer survivors (median age, 27.3 years; median follow-up, 19.8 years) diagnosed between 1963 and 2002.[26]
The cumulative incidence of developing a heart failure 40 years after a childhood cancer diagnosis was 4.4% (3.4%–5.5%) among cohort members.
The 20-year cumulative incidence of grade 3 or higher heart failure was greater among survivors treated in the more recent treatment periods (for survivors diagnosed between 1990 and 2001, 1.5%; between 1980 and 1989, 1.6%; and between 1970 and 1979, 0.5%) than in survivors treated earlier; however, mortality caused by heart failure decreased in the patients who were treated in the more recent periods.
Multivariable analysis demonstrated that patients who received higher doses of mitoxantrone or cyclophosphamide had an increased risk of heart failure compared with survivors who were exposed to lower doses.
The Pediatric Normal Tissue Effects in the Clinic (PENTEC) initiative reported on the risk of late cardiac disease for childhood cancer survivors who were treated with chemotherapy or radiation therapy.[27]
For each 10-Gy increase in corrected mean cardiac radiation dose in 1.8- to 2.0-Gy fractions, the estimated HRs were 2.01 (95% CI, 1.79–2.25) for coronary artery disease, 1.87 (95% CI, 1.70–2.06) for heart failure, 1.87 (95% CI, 1.78–1.96) for valvular disease, and 1.88 (95% CI, 1.75–2.03) for any cardiac disease.
For each 100-mg/m2 increase in cumulative anthracycline dose, the HR was 1.93 (95% CI, 1.58–2.36) for the development of heart failure, which is equivalent to an increase in mean heart radiation dose of approximately 10.5 Gy.
Treatment Risk Factors
Chemotherapy (in particular, anthracyclines and anthraquinones) and radiation therapy, both independently and in combination, increase the risk of cardiovascular disease in survivors of childhood cancer and are considered to be the most important risk factors contributing to premature cardiovascular disease in this population.[3,10,17,28]
Anthracyclines and related agents
Anthracyclines (e.g., doxorubicin, daunorubicin, idarubicin, and epirubicin) and anthraquinones (e.g., mitoxantrone) are known to directly injure cardiomyocytes through inhibition of topoisomerase 2-beta in cardiomyocytes and formation of reactive oxygen species, resulting in activation of cell-death pathways and inhibition of mitochondrial apoptosis.[29,30] The downstream results of cell death are changes in heart structure, including wall thinning, which leads to ventricular overload and pathological remodeling that, over time, leads to dysfunction and eventual clinical heart failure.[31,32]
Risk factors for anthracycline-related cardiomyopathy include the following:[19,33]
Cumulative dose, particularly greater than 250 to 300 mg/m2.
Younger age at time of exposure, particularly children younger than 5 years.
Increased time from exposure.
While there is no definitive safe lower dose threshold, doses in excess of 250 to 300 mg/m2 have been associated with a substantially increased risk of cardiomyopathy, with cumulative incidences exceeding 5% after 20 years of follow-up, and in some subgroups, reaching or exceeding 10% cumulative incidence by age 40 years.[18,19,32]
Concurrent chest or heart radiation therapy also further increases risk of cardiomyopathy,[10,28,34] as does the presence of other cardiometabolic traits such as hypertension.[8,35]
While development of clinical heart failure can occur within a few years after anthracycline exposure, in most survivors—even those who received very high doses—clinical manifestations may not occur for decades.
Anthracycline dose equivalency
Traditionally, anthracycline dose equivalence has largely been based on acute hematologic toxicity equivalence rather than late cardiac toxicity.
Analyses that pooled data from more than 28,000 long-term childhood cancer survivors monitored through age 40 years (resulting in 399 cardiomyopathy cases) have challenged previous assumptions that consider daunorubicin equivalent or nearly equivalent to doxorubicin.[36,37]
These investigations found that daunorubicin may be significantly less cardiotoxic than doxorubicin (equivalence ratio, 0.5; 95% CI, 0.4–0.7).[36]
Compared with doxorubicin, mitoxantrone may be significantly more cardiotoxic than previously thought (equivalence ratio, 10.5; 95% CI, 6.2–19.1), while epirubicin appeared to be doxorubicin isoequivalent (equivalence ratio, 0.8; 95% CI, 0.3–1.4).[37]
Data were too sparse to compare idarubicin with doxorubicin.
Anthracycline cardioprotection
Cardioprotective strategies that have been explored include the following:
New, less cardiotoxic agents and liposomal formulations. In general, data on whether liposomal formulations of anthracyclines reduce cardiac toxicity in children are limited.[38,39]
Prolonged infusion time. Prolonged infusion time has been associated with reduced heart failure in adult patients, but not in children.[40,41]
Concurrent administration of cardioprotectants. A variety of agents have been tested as cardioprotectants (amifostine, acetylcysteine, calcium channel blockers, carvedilol, coenzyme Q10, and L-carnitine), but none were beneficial and are not considered standard of care.[42,43]
Dexrazoxane. There are more data for dexrazoxane as a cardioprotectant, but mainly in adult patients with cancer. In particular, the U.S. Food and Drug Administration approved dexrazoxane for women with metastatic breast cancer who have received 300 mg/m2 of anthracyclines and who may benefit from further anthracycline-based therapy.[42]
Pediatric data show that dexrazoxane may ameliorate some markers of early cardiac toxicity for up to 5 years after therapy.[44–47]
Dexrazoxane may be associated with an increased risk of acute toxicities in some regimens.[48]
A long-term study assessed outcomes of newly diagnosed children with cancer who were treated with dexrazoxane-containing regimens in randomized clinical trials (cumulative prescribed doxorubicin dose, 100–360 mg/m2; median follow-up, 18.6 years). Dexrazoxane was not associated with cancer relapse (HR, 0.84; 95% CI, 0.63–1.13), second cancers (HR, 1.19; 95% CI, 0.62–2.30), all-cause mortality (HR, 1.07; 95% CI, 0.78–1.47), or cardiovascular mortality (HR, 1.45; 95% CI, 0.41–5.16).[49]
Another study evaluated long-term cardiac outcomes of 195 children with acute lymphoblastic leukemia or Hodgkin lymphoma who received doxorubicin with or without dexrazoxane in four randomized trials and patients with osteosarcoma who all received doxorubicin with dexrazoxane in a nonrandomized trial.[50]
At 18.1 years since cancer diagnosis (51% dexrazoxane exposed; cumulative doxorubicin dose, 297 mg/m2), dexrazoxane administration was associated with superior left ventricular fractional shortening (absolute difference, 1.4%; 95% CI, 0.3–2.5) and ejection fraction (absolute difference, 1.6%; 95% CI, 0.0–3.2) and lower myocardial stress per B-type natriuretic peptide (-6.7 pg/mL; 95% CI, -10.6 to -2.8).
Dexrazoxane use was associated with a reduced risk of lower left ventricular function (30% fractional shortening or 50% ejection fraction; OR, 0.24; 95% CI, 0.07–0.81), which was primarily observed in those treated with cumulative doxorubicin doses of 250 mg/m2 or higher.
Radiation therapy
While anthracyclines directly damage cardiomyocytes, radiation therapy primarily affects the fine vasculature of affected organs.[7]
Cardiovascular disease
Late effects of radiation therapy to the heart specifically include the following:
Delayed pericarditis, which can present abruptly or as a chronic pericardial effusion.
Pancarditis that includes pericardial and myocardial fibrosis, with or without endocardial fibroelastosis.
Cardiomyopathy (in the absence of significant pericardial disease), which can occur even without anthracycline exposure.
Ischemic heart disease.
Functional valve injury, often aortic.
Conduction defects.
These cardiac late effects are related to the following:
Individual radiation fraction size.
Volume of the heart that is exposed to radiation.[19,51]
Total radiation dose.
Various studies have demonstrated a substantially increased risk of these outcomes with higher radiation doses, particularly doses to the heart exceeding 35 Gy.[10,12,18,19,28]
At higher radiation doses, rates of heart failure, pericardial disease, and valvular disease have been reported to exceed 10% after 20 to 30 years. Although some studies suggest that doses less than 5 Gy may be associated with an increased risk of cardiovascular disease, the relative risk is small (i.e., 2.5) and the 95% CI is large (i.e., 0.2–41.5); moreover, the dosimetric analyses are generally estimations of incidental cardiac exposure.[10,18,28]
Low to moderate doses of radiation therapy (5.0–19.9 Gy) to large cardiac volumes (>50% of the heart) are associated with an increased rate of cardiac disease (i.e., 1.6-fold) compared with survivors who did not have any cardiac radiation therapy exposure.[19]
High doses of radiation (>20 Gy) to small cardiac volumes (0.1%–29.9%) are associated with an elevated rate of cardiac disease (relative rate, 2.4).[19,52]
Additional confirmatory data are needed for an accurate assessment of risk at very low cardiac doses.
Similar to anthracyclines, manifestation of these late effects may take years, if not decades, to present.
Patients who were exposed to both radiation therapy affecting the cardiovascular system and cardiotoxic chemotherapy agents are at even greater risk of late cardiovascular outcomes.[10,19] This risk may be decreasing based on Children’s Oncology Group (COG) Hodgkin lymphoma clinical trials spanning from 2002 to 2022.[53]
In a cohort study of 2,563 patients with Hodgkin lymphoma treated in four consecutive COG clinical trials between 2002 and 2022 (AHOD0031, AHOD0831, AHOD1331, S1826), the cumulative incidence of grades 3 to 5 cardiac disease was estimated to decrease, from 10% in the first trial to 6% in the last trial.
All patients received doxorubicin, 1,362 received mediastinal radiation therapy, and 307 received the cardioprotectant dexrazoxane (80% of children in the S1826 study). Radiation therapy was substantially reduced and refined, whereas the anthracycline doses were increased.
Based on modeling used as part of this study, survivors treated in the recent high-risk Hodgkin lymphoma trial (S1826), the 30-year risk of serious cardiac morbidity will be increased less than 2% above the expected rate in an untreated population (5%). Reductions in the proportion of children receiving mediastinal radiation therapy and increases in dexrazoxane use were estimated to offset the increased doxorubicin dose, producing a net reduction in late cardiac disease.
These results suggest that cardiac toxic effects are estimated to decrease from older to newer Hodgkin lymphoma trials, even in females aged 12 years and younger, who are most susceptible to anthracycline-induced heart failure.
Cerebrovascular disease
Cerebrovascular disease after radiation therapy exposure is another potential late effect observed in survivors.
Radiation-induced vascular damage is a complex process that involves both arterial and capillary damage, with veins being less sensitive.
The spectrum of abnormalities includes lacunar lesions, vascular malformations, telangiectasias, intracranial hemorrhage, moyamoya, microbleeds, and cavernomas, each with potential symptomatic consequences.[54,55]
While brain tumor survivors have traditionally had the greatest risk, other survivors exposed to cranial irradiation (≥18 Gy) and neck irradiation (≥40 Gy), such as leukemia and lymphoma survivors, have also been reported to be at increased risk.[56–59]
In a PENTEC analysis, risk of cerebrovascular toxicity and future risk of stroke were analyzed and modeled (COX proportional hazards and baseline cumulative incidences of stroke in nonradiation therapy exposed, age-matched cancer patients and the general population).[60]
Of 3,898 pediatric patients analyzed in five reports, 101 experienced at least one cerebrovascular toxicity (e.g., transient ischemic attack [TIA], stroke, moyamoya, or arteriopathy).
The risk of any cerebrovascular toxicity was 0.2% at 30 Gy, 1.3% at 45 Gy, and 4.4% at 54 Gy (D50, 75.6 Gy; 68.4–89.4 Gy).
At an attained age of 35 years, the predicted stroke incidence at d doses was 0.9% to 1.3% at 30 Gy, 1.8% to 2.7% at 45 Gy, and 2.8% to 4.1% at 54 Gy (population baseline risk, 0.2%–0.3%).
At an attained age of 45 years, the predicted stroke incidence was 2.1% to 4.2% at 30 Gy, 4.5% to 8.6% at 45 Gy, and 6.7% to 13.0% at 54 Gy (population baseline risk, 0.5%–1.0%).
Thus, the risk of future cerebrovascular toxicity continues to rise as follow-up duration increases. Additionally, because the stroke hazard was based on prescribed tumor bed dose rather than circle-of-Willis dose, the reported stroke risk was likely underestimated.
In lymphoma survivors who received only chest and/or neck radiation therapy, cerebrovascular disease is thought to be caused by large-vessel atherosclerosis and cardiac embolism.[57]
The risk increases with cumulative dose received. One study (N = 325) reported that the stroke hazard increased by 5% (HR, 1.05; 95% CI, 1.01–1.09; P = .02), with each 1 Gy increase in the radiation dose, leading to a cumulative incidence of 2% for the first stroke after 5 years and 4% after 10 years.[61]
Survivors who experienced a stroke were at much greater risk of recurrent strokes.[62]
Evidence (selected studies describing prevalence of and risk factors for cerebrovascular accident [CVA]/vascular disease):
The population-based British Childhood Cancer Survivor Study (n = 13,457) used Hospital Episode Statistics data for England to assess the risk of cerebrovascular-related hospitalizations (e.g., nontraumatic intracranial hemorrhage, cerebral infarction, or cerebral artery occlusion), particularly in patients older than 50 years.[59]
They found that 2.3% of the survivors had been hospitalized at least once for cerebrovascular disease, with a fourfold risk compared with the expected rate.
Survivors of central nervous system (CNS) tumors or leukemia who received cranial irradiation were at greatest risk of cerebrovascular disease (CNS tumor standardized hospitalization ratio [SHR], 15.6; 95% CI, 14.0–17.4 and leukemia SHR, 5.4; 95% CI, 4.5–6.4).
Beyond age 60 years, on average, 3.1% of CNS tumor survivors treated with cranial irradiation were hospitalized annually for cerebrovascular disease. By age 65 years, as many as 26% of these patients will have been hospitalized for a cerebrovascular event.
CCSS investigators observed that 295 of 13,060 participants (35% treated with cranial radiation therapy) reported having a stroke (6.3%; 95% CI, 5.1%–7.5%) by age 50 years (median follow-up, 19 years). For survivors considered to be high risk, the cumulative incidence of cerebrovascular disease up to age 50 was 19.9%.[63]
A retrospective study of 3,172 5-year survivors of childhood cancer monitored for a mean time of 26 years was formed from the Euro2K cohort, which included eight centers in France and the United Kingdom. Radiation doses to the circle of Willis were estimated for each of the 2,202 children who received radiation therapy.[64]
Patients who received radiation therapy had an 8.5-fold increased risk (95% CI, 6.3–11.0) of stroke in contrast to a nonelevated risk for patients who did not receive radiation therapy.
The relative risk was 15.7 (95% CI, 4.9–50.2) for doses of 40 Gy or higher.
At age 45 years, the cumulative incidence was 11.3% (95% CI, 7.1%–17.7%) in patients who received 10 Gy or higher to the circle of Willis, compared with 1% in the general population.
Investigators from the Teenage and Young Adult Cancer Survivor Study (N = 178,962) evaluated the risk of hospitalization for a cerebrovascular event among 5-year survivors of cancer diagnosed between age 15 and 39 years.[65]
The investigators found that survivors of adolescent and young adult cancers had a 40% increased risk of hospitalization for cerebrovascular event compared with the general population.
Survivors of CNS tumors (SHR, 4.6), head and neck tumors (SHR, 2.6), and leukemia (SHR, 2.5) had the highest risk of hospitalization for a cerebrovascular complication.
Males had significantly higher absolute excess risks than did females, especially among head and neck tumor survivors. By age 60 years, 9% of CNS tumor survivors, 6% of head and neck tumor survivors, and 5% of leukemia survivors had been hospitalized for a cerebrovascular event.
The risk of hospitalization for a cerebral infarction was particularly increased among survivors of a CNS tumor older than 60 years, whereas this risk was increased across all ages in survivors of head and neck tumors.
CCSS investigators evaluated the rates and predictors of recurrent stroke among participants who reported a first stroke.[62]
Among responding participants (329 of 443), 271 confirmed a first stroke (at median age, 19 years) and 70 reported a second stroke (at median age, 32 years).
Independent predictors of recurrent stroke included treatment with a cranial radiation therapy dose of 50 Gy or higher (vs. no cranial radiation therapy), history of hypertension, and age 40 years or older at first stroke (vs. age 0–17 years).
The 10-year cumulative incidence of late recurrent stroke was 21% overall and 33% for those treated with 50 Gy or higher of cranial radiation therapy.
A follow-up study of 224 CCSS participants who experienced stroke demonstrated an increased risk of all-cause and health-related mortality and a negative impact on social attainment, neurocognitive function, emotional distress, and other health-related quality-of-life measures.[66]
A multicenter retrospective Dutch study evaluated 5-year survivors of Hodgkin lymphoma diagnosed before age 51 years (25% pediatric-aged patients) who were followed for a median of 18 years. Among 2,201 survivors, 96 developed cerebrovascular disease (CVA and TIA).[57]
Most ischemic events were from large-artery atherosclerosis (36%) or cardiac embolism (24%).
The cumulative incidence of ischemic CVA or TIA 30 years after lymphoma treatment was 7%.
The overall standardized incidence ratio (SIR) was 2.2 for CVA and 3.1 for TIA. However, SIR estimates appeared to be greater among childhood cancer survivors, with SIRs of 3.8 for CVA and 7.6 for TIA.
Irradiation to the neck and mediastinum was an independent risk factor for ischemic cerebrovascular disease (HR, 2.5; 95% CI, 1.1–5.6) versus no radiation therapy. Treatment with chemotherapy was not associated with increased risk.
Hypertension, diabetes mellitus, and hypercholesterolemia were associated with the occurrence of ischemic cerebrovascular disease.
Venous thromboembolism
Children with cancer have an excess risk of venous thromboembolism within the first 5 years after diagnosis. However, the long-term risk of venous thromboembolism among childhood cancer survivors has not been well studied.[67]
CCSS investigators evaluated self-reported late-onset (5 or more years after cancer diagnosis) venous thromboembolism among cohort members (median follow-up, 21.3 years).[68]
The 35-year cumulative incidence of venous thromboembolism among survivors was 4.9%. This risk was twofold higher when compared with a sibling cohort (rate ratio, 2.2; 95% CI, 1.7–2.8).
Risk factors for venous thromboembolism among survivors included female sex, treatment with cisplatin or asparaginase, being affected by obesity or underweight, and recurrent primary or subsequent cancer.
The risk of late venous thromboembolism was higher among survivors of lower-extremity osteosarcoma treated with limb-sparing surgery compared with patients treated with amputation, possibly resulting from alterations in peripheral vascular anatomy and homeostasis.
Venous thromboembolism was associated with an almost-twofold increased risk of late mortality (RR, 1.9; 95% CI, 1.6–2.3).
Conventional cardiovascular conditions
Various cancer treatment exposures may also directly or indirectly influence the development of hypertension, diabetes mellitus, and dyslipidemia.
These conditions remain important among cancer survivors, as they do in the general population, in that they are independent risk factors in the development of cardiomyopathy, ischemic heart disease, and cerebrovascular disease.[8,69–72]
Childhood cancer survivors should be closely monitored for the development of these cardiovascular conditions because they represent potentially modifiable targets for intervention.
Related conditions such as obesity and various endocrinopathies (e.g., hypothyroidism, hypogonadism, growth hormone deficiency) that may be more common among subsets of childhood cancer survivors also need to be monitored. If these conditions are untreated/uncontrolled, they may be associated with a metabolic profile that increases cardiovascular risk.[9] For more information, see the Risk prediction for cardiovascular diseases section.
Other Risk Factors
Sex. Some, but not all, studies suggest that female sex may be associated with a greater risk of anthracycline-related cardiomyopathy.[7]
Genetics. There is emerging evidence that genetic factors, such as single nucleotide polymorphisms in genes regulating drug metabolism and distribution, could explain the heterogeneity in susceptibility to anthracycline-mediated cardiac injury.[73–81] However, these genetic findings still require additional validation before integration into any clinical screening algorithm.[81]
Peripartum Cardiac Dysfunction
Long-term survivors of childhood, adolescent, and young adult malignancies with past exposure to potentially cardiotoxic treatments are at risk of peripartum cardiac dysfunction.
In the general population, peripartum cardiomyopathy (PPCM) is a rare condition characterized by heart failure during pregnancy (usually the last trimester or <5 months postpartum). The estimated incidence in the general population is 1 case per 3,000 live births.[82]
There are limited data available about the prevalence in survivors of pediatric, adolescent, and young adult malignancies who have received cardiotoxic therapies.
In a retrospective series from SJCRH, 3 cases of peripartum cardiac dysfunction occurred in 1,554 completed pregnancies, which was an incidence of 0.2%; 27% of the 847 long-term survivors had not been exposed to cardiotoxic therapies.[83]
In a series of 64 women who had all received cardiotoxic therapy (44% received chest radiation therapy plus anthracyclines, 14% received chest radiation therapy, 42% received anthracycline alone), 5 women (7.8%) had peripartum cardiac events (3 symptomatic, 2 subclinical). Of the 110 live births, 2 involved PPCM, representing a 55-fold increased risk over the general population. Risk factors were younger age at cancer diagnosis and higher anthracycline dose. Postpartum cardiac function failed to return to baseline in four women (80%).[84]
A single-center study estimated the risk of developing congestive heart failure (CHF) during pregnancy as 1 out of 3 female cancer survivors with a history of cardiotoxicity.[85] Seventy-eight consecutive female cancer survivors previously exposed to potentially cardiotoxic treatments (chemotherapy and/or radiation therapy to the thorax as children, adolescents or young adults) who had 94 pregnancies were retrospectively identified through high-risk clinics between 2005 and 2015.
A total of 55 women received anthracyclines (range, 90–500 mg/m2), while 23 received nonanthracycline chemotherapy and/or radiation therapy only. Of the 13 women with prior cardiotoxicity, 8 had reduced left ventricular ejection fraction at the first antenatal visit. CHF occurred in five pregnancies (4 women; 5.3%).
The incidence of CHF was 31% for women with a history of cardiotoxicity and 0% for women without a history of cardiotoxicity. When comparing clinical characteristics between women with and without CHF, there was no difference in age of cancer diagnosis, cancer type, or exposure to anthracyclines.
Risk factors for developing CHF were history of cardiotoxicity before pregnancy, left ventricular systolic dysfunction at first antenatal visit, or receiving cardiac medications.
Based on available evidence about peripartum cardiomyopathy, the International Guideline Harmonization Group assessed that cardiomyopathy surveillance is reasonable before pregnancy or in the first trimester for female survivors of childhood, adolescent, and young adult cancer who are at moderate and high risk because they were treated with anthracyclines or chest radiation therapy.[33]
Mortality Risk After Major Cardiovascular Events
Survivors of childhood cancer represent a population at high risk of mortality after major cardiovascular events. Investigators estimated the cumulative incidence of all-cause and cardiovascular cause–specific mortality among survivors from the CCSS who had experienced a major cardiovascular event and compared them to siblings. They also compared the outcomes from the CCSS cancer survivors with a population-based cohort of racially diverse adults from the Coronary Artery Risk Development in Young Adults (CARDIA) study.[86]
Among the 25,658 childhood cancer survivors (median age at diagnosis, 7 years; median age at follow-up or death, 38 years) and 5,051 siblings, 1,780 survivors and 91 siblings experienced a cardiovascular event.
The 10-year all-cause mortality rate for survivors was 30% after heart failure, 36% after cardiovascular artery disease, and 29% after stroke. The 10-year all-cause mortality rate for siblings was 14% after heart failure, 14% after coronary artery disease, and 4% after stroke (P < .001 for all survivors).
All-cause mortality risks among childhood cancer survivors were increased after heart failure (HR, 7.32), coronary artery disease (HR, 5.54), and stroke (HR, 3.57).
Among 5,114 CARDIA participants, 345 had a major event. CARDIA participants were, on average, decades older at events (median age, 57 years vs. 31 years). However, mortality risks were similar, except that all-cause mortality after coronary artery disease was significantly increased among childhood cancer survivors (HR, 1.85).
Heart Transplant After Childhood Cancer
Data about the prevalence and outcomes of survivors with heart failure requiring heart transplant are limited.
In a study of solid organ transplants in 13,318 survivors in the CCSS, 62 survivors had end-stage heart failure that warranted heart transplants, 37 of whom received a heart transplant.[87]
At 35 years after cancer diagnosis, the cumulative incidence of heart transplant was 0.30%, and the cumulative incidence of being placed on the waiting list or receiving a heart was 0.49%.[87]
The 5-year survival rate from heart transplant was 80.6%, which is similar to the outcome in the general population of the same age range.[87]
Knowledge Deficits
While much knowledge has been gained over the past 20 years in better understanding the long-term burden and risk factors for cardiovascular disease among childhood cancer survivors, many areas of inquiry remain, and include the following:
Radiation may have both direct and indirect effects on vascular endothelium, contributing to vascular damage beyond the primary radiation field.[88]
The long-term effects of lower radiation doses, particularly in the setting of advanced technology that allows tumor targeting from multiple directions and reduces exposure to surrounding normal tissues, remain to be determined.[89]
The long-term effects of many newer anticancer agents that are based on molecular targets remain unclear, although some of them are known to have shorter-term cardiac toxicity.[16]
The efficacy of cardioprotective strategies, including the use of alternative anthracycline formulations that appear promising in adults, requires further study in children.[43]
Screening, Surveillance, and Counseling
The International Guideline Harmonization Group has worked collaboratively to harmonize evidence-based cardiac surveillance recommendations and have identified knowledge deficits to help guide future studies.[33,72] Risk groups defined by cumulative exposures of anthracycline and chest-directed radiation therapy as well as cardiomyopathy surveillance recommendations are summarized in Table 2.
Consensus regarding evidence about screening, surveillance, and counseling
There is no clear evidence (at least through age 50 years or 30–40 years posttreatment) that a plateau in risk occurs after a certain time among survivors exposed to cancer treatments associated with cardiovascular late effects.[90,91] Thus, life-long surveillance is recommended by one group, even if the cost-effectiveness of certain screening strategies remains unclear.[33,92–94]
Table 2. Risk Groups and Cardiomyopathy Surveillance Recommendations for Survivors of Childhood, Adolescent, and Young Adult Cancera
A growing body of literature is beginning to establish the yield from these screening studies, which will help inform future guidelines.[9,95–97] In these studies, for example, among adult-aged survivors of childhood cancer, evidence for cardiomyopathy on the basis of echocardiographic changes was found in approximately 6% of at-risk survivors. Overall, in a cohort of more than 1,000 survivors (median age, 32 years), nearly 60% of screened at-risk survivors had some clinically ascertained cardiac abnormality identified.[9]
Given the growing evidence that conventional cardiovascular conditions such as hypertension, dyslipidemia, and diabetes substantially increase the risk of more serious cardiovascular disease among survivors, clinicians should carefully consider baseline and follow-up screening and treatment of these comorbid conditions that impact cardiovascular health (see Table 3).[8,57,69,98]
There is also emerging evidence that adoption of healthier lifestyle factors may decrease future cardiovascular morbidity in at-risk survivors.[99,100] Thus, similar to the general population, survivors should be counseled about maintaining a healthy weight, participating in regular physical activity, adhering to a heart-healthy diet, and abstaining from smoking.
The COG has organized handouts on cardiovascular disease and related topics, including lifestyle choices, written for a lay audience to facilitate counseling and education of survivors. For more information, see the COG Survivorship Guidelines.
Predicting Cardiovascular Disease Risk
Attempts to develop more individualized risk prediction for cardiovascular disease may help refine surveillance and counseling in the future.
Several groups have collaborated to develop and validate individualized risk calculators for heart failure, ischemic heart disease, and stroke through age 50 years.[34,63,98]
Updated models based only on CCSS data have incorporated hypertension, dyslipidemia, and diabetes status across time to further refine prediction.[98]
An online risk calculator incorporating these models is available on the CCSS website.
Risk prediction and interventions for cardiovascular diseases
Using data from four large, well-annotated childhood cancer survivor cohorts (CCSS, National Wilms Tumor Study Group, the Netherlands, and SJCRH), a heart failure risk calculator based on readily available demographic and treatment characteristics has been created and validated. This calculator may provide more individualized clinical heart failure risk estimation for 5-year survivors of childhood cancer who have recently completed therapy, through age 40 years. Because of the young age of participants at the time of baseline prediction (5-year survival), this estimator is limited in that information on conventional cardiovascular conditions such as hypertension, dyslipidemia, or diabetes could not be incorporated.[34]
In another collaborative study, data from the CCSS, Netherlands, and SJCRH were used to develop risk-prediction models for ischemic heart disease and stroke among 5-year survivors of childhood cancer through age 50 years. Risk scores derived from a standard prediction model that included sex, chemotherapy exposure, and radiation therapy exposure identified statistically distinct low-risk, moderate-risk, and high-risk groups. The cumulative incidences at age 50 years among CCSS low-risk groups were less than 5%, compared with approximately 20% for high-risk groups and only 1% for siblings.[63]
Traditional cardiovascular risk factors remain important for predicting risk of cardiovascular disease among adult-aged survivors of childhood cancer. This finding was demonstrated by a CCSS investigation that constructed prediction models accounting for cardiotoxic cancer treatment exposures, combined with information on traditional cardiovascular risk factors such as hypertension, dyslipidemia, and diabetes. Risk scores based on demographic, cancer treatment, hypertension, dyslipidemia, and diabetes information showed good performance (area under the receiver operating characteristic curve and concordance statistics ≥0.70) for predicting cardiovascular events in the models applied to the discovery and replication cohorts. The most influential exposures were anthracycline chemotherapy, radiation therapy, diabetes, and hypertension.[98]
Physical activity has been shown to be a safe intervention against the high burden of cardiovascular late effects in childhood cancer survivors. A randomized controlled trial investigated the effect of a partially supervised, personalized, 1-year physical activity intervention on cardiovascular health in long-term survivors of childhood cancer.[101] There were 151 childhood cancer survivors randomly assigned (1:1) to either perform more than 2.5 hours of additional intense physical activity per week (intervention group) or continue exercise as usual (control group). A significant and robust reduction of the cardiovascular disease risk score was noted at 6 months and 12 months in the intervention group, compared with the control group. The difference in the reduction of the cardiovascular disease risk z-score of -0.18 (P = .003) at 12 months favored the intervention group.
Table 3. Cardiovascular Late Effectsa,b
Predisposing Therapy
Potential Cardiovascular Effects
Health Screening
aThe Children’s Oncology Group (COG) guidelines also cover other conditions that may influence cardiovascular risk, such as obesity and diabetes mellitus/impaired glucose metabolism.
Fasting lipid profile and diabetes screening every 2 years
Heavy metals (carboplatin, cisplatin), and ifosfamide exposure; radiation exposing the kidneys; HSCT; nephrectomy
Hypertension (from renal toxicity)
Yearly blood pressure test; renal function laboratory studies at entry into long-term follow-up and repeat as clinically indicated
HSCT = hematopoietic stem cell transplant.
References
Armstrong GT, Chen Y, Yasui Y, et al.: Reduction in Late Mortality among 5-Year Survivors of Childhood Cancer. N Engl J Med 374 (9): 833-42, 2016. [PUBMED Abstract]
Fidler MM, Reulen RC, Winter DL, et al.: Long term cause specific mortality among 34 489 five year survivors of childhood cancer in Great Britain: population based cohort study. BMJ 354: i4351, 2016. [PUBMED Abstract]
Henson KE, Reulen RC, Winter DL, et al.: Cardiac Mortality Among 200 000 Five-Year Survivors of Cancer Diagnosed at 15 to 39 Years of Age: The Teenage and Young Adult Cancer Survivor Study. Circulation 134 (20): 1519-1531, 2016. [PUBMED Abstract]
Suh E, Stratton KL, Leisenring WM, et al.: Late mortality and chronic health conditions in long-term survivors of early-adolescent and young adult cancers: a retrospective cohort analysis from the Childhood Cancer Survivor Study. Lancet Oncol 21 (3): 421-435, 2020. [PUBMED Abstract]
Hammoud RA, Liu Q, Dixon SB, et al.: The burden of cardiovascular disease and risk for subsequent major adverse cardiovascular events in survivors of childhood cancer: a prospective, longitudinal analysis from the St Jude Lifetime Cohort Study. Lancet Oncol 25 (6): 811-822, 2024. [PUBMED Abstract]
Fidler MM, Reulen RC, Henson K, et al.: Population-Based Long-Term Cardiac-Specific Mortality Among 34 489 Five-Year Survivors of Childhood Cancer in Great Britain. Circulation 135 (10): 951-963, 2017. [PUBMED Abstract]
Bansal N, Blanco JG, Sharma UC, et al.: Cardiovascular diseases in survivors of childhood cancer. Cancer Metastasis Rev 39 (1): 55-68, 2020. [PUBMED Abstract]
Armstrong GT, Oeffinger KC, Chen Y, et al.: Modifiable risk factors and major cardiac events among adult survivors of childhood cancer. J Clin Oncol 31 (29): 3673-80, 2013. [PUBMED Abstract]
Hudson MM, Ness KK, Gurney JG, et al.: Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA 309 (22): 2371-81, 2013. [PUBMED Abstract]
Haddy N, Diallo S, El-Fayech C, et al.: Cardiac Diseases Following Childhood Cancer Treatment: Cohort Study. Circulation 133 (1): 31-8, 2016. [PUBMED Abstract]
Cotton CA, Peterson S, Norkool PA, et al.: Early and late mortality after diagnosis of wilms tumor. J Clin Oncol 27 (8): 1304-9, 2009. [PUBMED Abstract]
Schellong G, Riepenhausen M, Bruch C, et al.: Late valvular and other cardiac diseases after different doses of mediastinal radiotherapy for Hodgkin disease in children and adolescents: report from the longitudinal GPOH follow-up project of the German-Austrian DAL-HD studies. Pediatr Blood Cancer 55 (6): 1145-52, 2010. [PUBMED Abstract]
Green DM, Kun LE, Matthay KK, et al.: Relevance of historical therapeutic approaches to the contemporary treatment of pediatric solid tumors. Pediatr Blood Cancer 60 (7): 1083-94, 2013. [PUBMED Abstract]
Hudson MM, Neglia JP, Woods WG, et al.: Lessons from the past: opportunities to improve childhood cancer survivor care through outcomes investigations of historical therapeutic approaches for pediatric hematological malignancies. Pediatr Blood Cancer 58 (3): 334-43, 2012. [PUBMED Abstract]
de Baat EC, van Dalen EC, Mulder RL, et al.: Primary cardioprotection with dexrazoxane in patients with childhood cancer who are expected to receive anthracyclines: recommendations from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Child Adolesc Health 6 (12): 885-894, 2022. [PUBMED Abstract]
Chow EJ, Antal Z, Constine LS, et al.: New Agents, Emerging Late Effects, and the Development of Precision Survivorship. J Clin Oncol 36 (21): 2231-2240, 2018. [PUBMED Abstract]
de Baat EC, Feijen EAM, Reulen RC, et al.: Risk Factors for Heart Failure Among Pan-European Childhood Cancer Survivors: A PanCareSurFup and ProCardio Cohort and Nested Case-Control Study. J Clin Oncol 41 (1): 96-106, 2023. [PUBMED Abstract]
Mulrooney DA, Hyun G, Ness KK, et al.: Major cardiac events for adult survivors of childhood cancer diagnosed between 1970 and 1999: report from the Childhood Cancer Survivor Study cohort. BMJ 368: l6794, 2020. [PUBMED Abstract]
Bates JE, Howell RM, Liu Q, et al.: Therapy-Related Cardiac Risk in Childhood Cancer Survivors: An Analysis of the Childhood Cancer Survivor Study. J Clin Oncol 37 (13): 1090-1101, 2019. [PUBMED Abstract]
Bates JE, Shrestha S, Liu Q, et al.: Cardiac Substructure Radiation Dose and Risk of Late Cardiac Disease in Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (22): 3826-3838, 2023. [PUBMED Abstract]
Chow EJ, Chen Y, Armstrong GT, et al.: Underdiagnosis and Undertreatment of Modifiable Cardiovascular Risk Factors Among Survivors of Childhood Cancer. J Am Heart Assoc 11 (12): e024735, 2022. [PUBMED Abstract]
Goldberg JF, Hyun G, Ness KK, et al.: Dyslipidemia and cardiovascular disease among childhood cancer survivors: a St. Jude Lifetime Cohort report. J Natl Cancer Inst 116 (3): 408-420, 2024. [PUBMED Abstract]
Kooijmans ECM, van der Pal HJH, Pluijm SMF, et al.: Hypertension in long-term childhood cancer survivors after treatment with potentially nephrotoxic therapy; DCCSS-LATER 2: Renal study. Eur J Cancer 172: 287-299, 2022. [PUBMED Abstract]
Bhakta N, Liu Q, Yeo F, et al.: Cumulative burden of cardiovascular morbidity in paediatric, adolescent, and young adult survivors of Hodgkin’s lymphoma: an analysis from the St Jude Lifetime Cohort Study. Lancet Oncol 17 (9): 1325-34, 2016. [PUBMED Abstract]
Mulrooney DA, Soliman EZ, Ehrhardt MJ, et al.: Electrocardiographic abnormalities and mortality in aging survivors of childhood cancer: A report from the St Jude Lifetime Cohort Study. Am Heart J 189: 19-27, 2017. [PUBMED Abstract]
Feijen EAML, Font-Gonzalez A, Van der Pal HJH, et al.: Risk and Temporal Changes of Heart Failure Among 5-Year Childhood Cancer Survivors: a DCOG-LATER Study. J Am Heart Assoc 8 (1): e009122, 2019. [PUBMED Abstract]
Bates JE, Rancati T, Keshavarz H, et al.: Cardiac Disease in Childhood Cancer Survivors Treated With Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 522-532, 2024. [PUBMED Abstract]
van der Pal HJ, van Dalen EC, van Delden E, et al.: High risk of symptomatic cardiac events in childhood cancer survivors. J Clin Oncol 30 (13): 1429-37, 2012. [PUBMED Abstract]
Zhang S, Liu X, Bawa-Khalfe T, et al.: Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 18 (11): 1639-42, 2012. [PUBMED Abstract]
Lyu YL, Kerrigan JE, Lin CP, et al.: Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 67 (18): 8839-46, 2007. [PUBMED Abstract]
Hudson MM, Rai SN, Nunez C, et al.: Noninvasive evaluation of late anthracycline cardiac toxicity in childhood cancer survivors. J Clin Oncol 25 (24): 3635-43, 2007. [PUBMED Abstract]
van der Pal HJ, van Dalen EC, Hauptmann M, et al.: Cardiac function in 5-year survivors of childhood cancer: a long-term follow-up study. Arch Intern Med 170 (14): 1247-55, 2010. [PUBMED Abstract]
Ehrhardt MJ, Leerink JM, Mulder RL, et al.: Systematic review and updated recommendations for cardiomyopathy surveillance for survivors of childhood, adolescent, and young adult cancer from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol 24 (3): e108-e120, 2023. [PUBMED Abstract]
Chow EJ, Chen Y, Kremer LC, et al.: Individual prediction of heart failure among childhood cancer survivors. J Clin Oncol 33 (5): 394-402, 2015. [PUBMED Abstract]
Armenian SH, Sun CL, Vase T, et al.: Cardiovascular risk factors in hematopoietic cell transplantation survivors: role in development of subsequent cardiovascular disease. Blood 120 (23): 4505-12, 2012. [PUBMED Abstract]
Feijen EA, Leisenring WM, Stratton KL, et al.: Equivalence Ratio for Daunorubicin to Doxorubicin in Relation to Late Heart Failure in Survivors of Childhood Cancer. J Clin Oncol 33 (32): 3774-80, 2015. [PUBMED Abstract]
Feijen EAM, Leisenring WM, Stratton KL, et al.: Derivation of Anthracycline and Anthraquinone Equivalence Ratios to Doxorubicin for Late-Onset Cardiotoxicity. JAMA Oncol 5 (6): 864-871, 2019. [PUBMED Abstract]
van Dalen EC, Michiels EM, Caron HN, et al.: Different anthracycline derivates for reducing cardiotoxicity in cancer patients. Cochrane Database Syst Rev (5): CD005006, 2010. [PUBMED Abstract]
Krauss AC, Gao X, Li L, et al.: FDA Approval Summary: (Daunorubicin and Cytarabine) Liposome for Injection for the Treatment of Adults with High-Risk Acute Myeloid Leukemia. Clin Cancer Res 25 (9): 2685-2690, 2019. [PUBMED Abstract]
van Dalen EC, van der Pal HJ, Caron HN, et al.: Different dosage schedules for reducing cardiotoxicity in cancer patients receiving anthracycline chemotherapy. Cochrane Database Syst Rev (4): CD005008, 2009. [PUBMED Abstract]
Lipshultz SE, Giantris AL, Lipsitz SR, et al.: Doxorubicin administration by continuous infusion is not cardioprotective: the Dana-Farber 91-01 Acute Lymphoblastic Leukemia protocol. J Clin Oncol 20 (6): 1677-82, 2002. [PUBMED Abstract]
Hensley ML, Hagerty KL, Kewalramani T, et al.: American Society of Clinical Oncology 2008 clinical practice guideline update: use of chemotherapy and radiation therapy protectants. J Clin Oncol 27 (1): 127-45, 2009. [PUBMED Abstract]
van Dalen EC, Caron HN, Dickinson HO, et al.: Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst Rev (6): CD003917, 2011. [PUBMED Abstract]
Wexler LH, Andrich MP, Venzon D, et al.: Randomized trial of the cardioprotective agent ICRF-187 in pediatric sarcoma patients treated with doxorubicin. J Clin Oncol 14 (2): 362-72, 1996. [PUBMED Abstract]
Lipshultz SE, Scully RE, Lipsitz SR, et al.: Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol 11 (10): 950-61, 2010. [PUBMED Abstract]
Asselin BL, Devidas M, Chen L, et al.: Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children’s Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol 34 (8): 854-62, 2016. [PUBMED Abstract]
Shaikh F, Dupuis LL, Alexander S, et al.: Cardioprotection and Second Malignant Neoplasms Associated With Dexrazoxane in Children Receiving Anthracycline Chemotherapy: A Systematic Review and Meta-Analysis. J Natl Cancer Inst 108 (4): , 2016. [PUBMED Abstract]
Schwartz CL, Constine LS, Villaluna D, et al.: A risk-adapted, response-based approach using ABVE-PC for children and adolescents with intermediate- and high-risk Hodgkin lymphoma: the results of P9425. Blood 114 (10): 2051-9, 2009. [PUBMED Abstract]
Chow EJ, Aplenc R, Vrooman LM, et al.: Late health outcomes after dexrazoxane treatment: A report from the Children’s Oncology Group. Cancer 128 (4): 788-796, 2022. [PUBMED Abstract]
Chow EJ, Aggarwal S, Doody DR, et al.: Dexrazoxane and Long-Term Heart Function in Survivors of Childhood Cancer. J Clin Oncol 41 (12): 2248-2257, 2023. [PUBMED Abstract]
Mansouri I, Allodji RS, Hill C, et al.: The role of irradiated heart and left ventricular volumes in heart failure occurrence after childhood cancer. Eur J Heart Fail 21 (4): 509-518, 2019. [PUBMED Abstract]
Shrestha S, Bates JE, Liu Q, et al.: Radiation therapy related cardiac disease risk in childhood cancer survivors: Updated dosimetry analysis from the Childhood Cancer Survivor Study. Radiother Oncol 163: 199-208, 2021. [PUBMED Abstract]
Lo AC, Liu A, Liu Q, et al.: Late Cardiac Toxic Effects Associated With Treatment Protocols for Hodgkin Lymphoma in Children. JAMA Netw Open 7 (1): e2351062, 2024. [PUBMED Abstract]
Passos J, Nzwalo H, Valente M, et al.: Microbleeds and cavernomas after radiotherapy for paediatric primary brain tumours. J Neurol Sci 372: 413-416, 2017. [PUBMED Abstract]
Bowers DC, Liu Y, Leisenring W, et al.: Late-occurring stroke among long-term survivors of childhood leukemia and brain tumors: a report from the Childhood Cancer Survivor Study. J Clin Oncol 24 (33): 5277-82, 2006. [PUBMED Abstract]
De Bruin ML, Dorresteijn LD, van’t Veer MB, et al.: Increased risk of stroke and transient ischemic attack in 5-year survivors of Hodgkin lymphoma. J Natl Cancer Inst 101 (13): 928-37, 2009. [PUBMED Abstract]
van Dijk IW, van der Pal HJ, van Os RM, et al.: Risk of Symptomatic Stroke After Radiation Therapy for Childhood Cancer: A Long-Term Follow-Up Cohort Analysis. Int J Radiat Oncol Biol Phys 96 (3): 597-605, 2016. [PUBMED Abstract]
Reulen RC, Guha J, Bright CJ, et al.: Risk of cerebrovascular disease among 13 457 five-year survivors of childhood cancer: A population-based cohort study. Int J Cancer 148 (3): 572-583, 2021. [PUBMED Abstract]
Waxer JF, Wong K, Modiri A, et al.: Risk of Cerebrovascular Events Among Childhood and Adolescent Patients Receiving Cranial Radiation Therapy: A PENTEC Normal Tissue Outcomes Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 417-430, 2024. [PUBMED Abstract]
Mueller S, Sear K, Hills NK, et al.: Risk of first and recurrent stroke in childhood cancer survivors treated with cranial and cervical radiation therapy. Int J Radiat Oncol Biol Phys 86 (4): 643-8, 2013. [PUBMED Abstract]
Fullerton HJ, Stratton K, Mueller S, et al.: Recurrent stroke in childhood cancer survivors. Neurology 85 (12): 1056-64, 2015. [PUBMED Abstract]
Chow EJ, Chen Y, Hudson MM, et al.: Prediction of Ischemic Heart Disease and Stroke in Survivors of Childhood Cancer. J Clin Oncol 36 (1): 44-52, 2018. [PUBMED Abstract]
El-Fayech C, Haddy N, Allodji RS, et al.: Cerebrovascular Diseases in Childhood Cancer Survivors: Role of the Radiation Dose to Willis Circle Arteries. Int J Radiat Oncol Biol Phys 97 (2): 278-286, 2017. [PUBMED Abstract]
Bright CJ, Hawkins MM, Guha J, et al.: Risk of Cerebrovascular Events in 178 962 Five-Year Survivors of Cancer Diagnosed at 15 to 39 Years of Age: The TYACSS (Teenage and Young Adult Cancer Survivor Study). Circulation 135 (13): 1194-1210, 2017. [PUBMED Abstract]
Mueller S, Kline CN, Buerki RA, et al.: Stroke impact on mortality and psychologic morbidity within the Childhood Cancer Survivor Study. Cancer 126 (5): 1051-1059, 2020. [PUBMED Abstract]
Walker AJ, Grainge MJ, Card TR, et al.: Venous thromboembolism in children with cancer – a population-based cohort study. Thromb Res 133 (3): 340-4, 2014. [PUBMED Abstract]
Madenci AL, Weil BR, Liu Q, et al.: Long-Term Risk of Venous Thromboembolism in Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol : JCO2018784595, 2018. [PUBMED Abstract]
Mueller S, Fullerton HJ, Stratton K, et al.: Radiation, atherosclerotic risk factors, and stroke risk in survivors of pediatric cancer: a report from the Childhood Cancer Survivor Study. Int J Radiat Oncol Biol Phys 86 (4): 649-55, 2013. [PUBMED Abstract]
Chao C, Xu L, Bhatia S, et al.: Cardiovascular Disease Risk Profiles in Survivors of Adolescent and Young Adult (AYA) Cancer: The Kaiser Permanente AYA Cancer Survivors Study. J Clin Oncol 34 (14): 1626-33, 2016. [PUBMED Abstract]
Winther JF, Bhatia S, Cederkvist L, et al.: Risk of cardiovascular disease among Nordic childhood cancer survivors with diabetes mellitus: A report from adult life after childhood cancer in Scandinavia. Cancer 124 (22): 4393-4400, 2018. [PUBMED Abstract]
van Dalen EC, Mulder RL, Suh E, et al.: Coronary artery disease surveillance among childhood, adolescent and young adult cancer survivors: A systematic review and recommendations from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Eur J Cancer 156: 127-137, 2021. [PUBMED Abstract]
Lipshultz SE, Lipsitz SR, Kutok JL, et al.: Impact of hemochromatosis gene mutations on cardiac status in doxorubicin-treated survivors of childhood high-risk leukemia. Cancer 119 (19): 3555-62, 2013. [PUBMED Abstract]
Visscher H, Ross CJ, Rassekh SR, et al.: Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatr Blood Cancer 60 (8): 1375-81, 2013. [PUBMED Abstract]
Wang X, Liu W, Sun CL, et al.: Hyaluronan synthase 3 variant and anthracycline-related cardiomyopathy: a report from the children’s oncology group. J Clin Oncol 32 (7): 647-53, 2014. [PUBMED Abstract]
Wang X, Sun CL, Quiñones-Lombraña A, et al.: CELF4 Variant and Anthracycline-Related Cardiomyopathy: A Children’s Oncology Group Genome-Wide Association Study. J Clin Oncol 34 (8): 863-70, 2016. [PUBMED Abstract]
Aminkeng F, Bhavsar AP, Visscher H, et al.: A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat Genet 47 (9): 1079-84, 2015. [PUBMED Abstract]
Visscher H, Rassekh SR, Sandor GS, et al.: Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics 16 (10): 1065-76, 2015. [PUBMED Abstract]
Singh P, Wang X, Hageman L, et al.: Association of GSTM1 null variant with anthracycline-related cardiomyopathy after childhood cancer-A Children’s Oncology Group ALTE03N1 report. Cancer 126 (17): 4051-4058, 2020. [PUBMED Abstract]
Sapkota Y, Ehrhardt MJ, Qin N, et al.: A Novel Locus on 6p21.2 for Cancer Treatment-Induced Cardiac Dysfunction Among Childhood Cancer Survivors. J Natl Cancer Inst 114 (8): 1109-1116, 2022. [PUBMED Abstract]
Davies SM: Getting to the heart of the matter. J Clin Oncol 30 (13): 1399-400, 2012. [PUBMED Abstract]
Hines MR, Mulrooney DA, Hudson MM, et al.: Pregnancy-associated cardiomyopathy in survivors of childhood cancer. J Cancer Surviv 10 (1): 113-21, 2016. [PUBMED Abstract]
Chait-Rubinek L, Mariani JA, Goroncy N, et al.: A Retrospective Evaluation of Risk of Peripartum Cardiac Dysfunction in Survivors of Childhood, Adolescent and Young Adult Malignancies. Cancers (Basel) 11 (8): , 2019. [PUBMED Abstract]
Liu S, Aghel N, Belford L, et al.: Cardiac Outcomes in Pregnant Women With Treated Cancer. J Am Coll Cardiol 72 (17): 2087-2089, 2018. [PUBMED Abstract]
Bottinor W, Im C, Doody DR, et al.: Mortality After Major Cardiovascular Events in Survivors of Childhood Cancer. J Am Coll Cardiol 83 (8): 827-838, 2024. [PUBMED Abstract]
Dietz AC, Seidel K, Leisenring WM, et al.: Solid organ transplantation after treatment for childhood cancer: a retrospective cohort analysis from the Childhood Cancer Survivor Study. Lancet Oncol 20 (10): 1420-1431, 2019. [PUBMED Abstract]
Brouwer CA, Postma A, Hooimeijer HL, et al.: Endothelial damage in long-term survivors of childhood cancer. J Clin Oncol 31 (31): 3906-13, 2013. [PUBMED Abstract]
Maraldo MV, Jørgensen M, Brodin NP, et al.: The impact of involved node, involved field and mantle field radiotherapy on estimated radiation doses and risk of late effects for pediatric patients with Hodgkin lymphoma. Pediatr Blood Cancer 61 (4): 717-22, 2014. [PUBMED Abstract]
Tukenova M, Guibout C, Oberlin O, et al.: Role of cancer treatment in long-term overall and cardiovascular mortality after childhood cancer. J Clin Oncol 28 (8): 1308-15, 2010. [PUBMED Abstract]
Armstrong GT, Kawashima T, Leisenring W, et al.: Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol 32 (12): 1218-27, 2014. [PUBMED Abstract]
Chen AB, Punglia RS, Kuntz KM, et al.: Cost effectiveness and screening interval of lipid screening in Hodgkin’s lymphoma survivors. J Clin Oncol 27 (32): 5383-9, 2009. [PUBMED Abstract]
Wong FL, Bhatia S, Landier W, et al.: Cost-effectiveness of the children’s oncology group long-term follow-up screening guidelines for childhood cancer survivors at risk for treatment-related heart failure. Ann Intern Med 160 (10): 672-83, 2014. [PUBMED Abstract]
Yeh JM, Nohria A, Diller L: Routine echocardiography screening for asymptomatic left ventricular dysfunction in childhood cancer survivors: a model-based estimation of the clinical and economic effects. Ann Intern Med 160 (10): 661-71, 2014. [PUBMED Abstract]
Landier W, Armenian SH, Lee J, et al.: Yield of screening for long-term complications using the children’s oncology group long-term follow-up guidelines. J Clin Oncol 30 (35): 4401-8, 2012. [PUBMED Abstract]
Ramjaun A, AlDuhaiby E, Ahmed S, et al.: Echocardiographic Detection of Cardiac Dysfunction in Childhood Cancer Survivors: How Long Is Screening Required? Pediatr Blood Cancer 62 (12): 2197-203, 2015. [PUBMED Abstract]
Spewak MB, Williamson RS, Mertens AC, et al.: Yield of screening echocardiograms during pediatric follow-up in survivors treated with anthracyclines and cardiotoxic radiation. Pediatr Blood Cancer 64 (6): , 2017. [PUBMED Abstract]
Chen Y, Chow EJ, Oeffinger KC, et al.: Traditional Cardiovascular Risk Factors and Individual Prediction of Cardiovascular Events in Childhood Cancer Survivors. J Natl Cancer Inst 112 (3): 256-265, 2020. [PUBMED Abstract]
Scott JM, Li N, Liu Q, et al.: Association of Exercise With Mortality in Adult Survivors of Childhood Cancer. JAMA Oncol 4 (10): 1352-1358, 2018. [PUBMED Abstract]
Schindera C, Zürcher SJ, Jung R, et al.: Physical fitness and modifiable cardiovascular disease risk factors in survivors of childhood cancer: A report from the SURfit study. Cancer 127 (10): 1690-1698, 2021. [PUBMED Abstract]
Rueegg CS, Zürcher SJ, Schindera C, et al.: Effect of a 1-year physical activity intervention on cardiovascular health in long-term childhood cancer survivors-a randomised controlled trial (SURfit). Br J Cancer 129 (8): 1284-1297, 2023. [PUBMED Abstract]
Late Effects of the Central Nervous System
Neurocognitive
Neurocognitive late effects are commonly observed after treatment of malignancies that require central nervous system (CNS)–directed therapies, including the following:
Cranial radiation therapy.
Systemic therapy with high-dose methotrexate or cytarabine.
Children with CNS tumors or acute lymphoblastic leukemia (ALL) are most likely to be affected. Risk factors for the development of neurocognitive late effects include the following:[3–7]
Female sex.
Younger age at the time of treatment.
Tumor location.
Treatment with cranial radiation therapy and/or chemotherapeutic agents (systemic or intrathecal).
Higher cranial radiation dose.
Time since treatment.
Cognitive phenotypes observed in childhood survivors of ALL and CNS tumors may differ from traditional developmental disorders. For example, the phenotype of attention problems in ALL and brain tumor survivors appears to differ from developmental attention-deficit/hyperactivity disorder (ADHD) in that few survivors demonstrate significant hyperactivity/impulsivity, but instead have associated difficulties with processing speed and executive function.[8,9]
A Pediatric Normal Tissue Effects in the Clinic (PENTEC) comprehensive review was performed to develop models to facilitate the identification of dose constraints for radiation-associated CNS morbidities.[10]
Models suggest a 5% risk of a subsequent intelligence quotient (IQ) lower than 85 when the following occurs:
10% of the brain is irradiated to 35.7 Gy.
20% of the brain is irradiated to 29.1 Gy.
50% of the brain is irradiated to 22.2 Gy.
100% of the brain is irradiated to 18.1 Gy.
Note: All at 2 Gy/fraction and without methotrexate.
Methotrexate exposure increased the risk of an IQ score lower than 85, similar to a generalized uniform brain radiation dose of 5.9 Gy. However, a limitation to this analysis was a lack of data about the total methotrexate doses, both intravenous and intrathecal.
The model for predicting expected IQ, which also included the effect of dose, age, and methotrexate, suggested that each of these factors has an independent but probably cumulative effect on IQ.
In addition to the direct effects of neurotoxic therapies like cranial radiation, Childhood Cancer Survivor Study (CCSS) investigators observed that chronic health conditions resulting from non-neurotoxic treatment exposures (e.g., thoracic radiation) can adversely impact neurocognitive function presumably mediated by chronic cardiopulmonary and endocrine dysfunction.[11] In addition, some sequelae of neurotoxic therapy (e.g., severe hearing loss) have been associated with neurocognitive deficits independent of the neurotoxic treatment received.[12]
A related investigation from the CCSS evaluated longitudinal associations between physical activity and neurocognitive problems in adult survivors of childhood cancer.[13]
Survivors were less likely than their siblings to report consistent physical activity (28.1% vs. 33.6%).
Survivors who reported more consistent physical activity had fewer neurocognitive problems and larger improvements in cognitive concerns years after treatment.
Body mass index (BMI) and severe chronic health conditions partially mediated the physical activity–neurocognitive associations, but the mediation effects were small.
A subsequent systematic review and meta-analysis compared the effects of physical activity or exercise interventions on cognitive function among individuals diagnosed with cancer (aged 0–19 years) with that of controls. Twenty-two unique studies (16 randomized controlled trials) were found with data on 12,767 individuals.[14] The median age at the start of the study was 12 years (interquartile range [IQR], 11–14 years), the median time from the end of cancer treatment was 2.5 years (IQR, 1.1–3.0 years), and the median intervention period was 12 weeks (IQR, 10–24 weeks).
Compared with controls, there was moderate-quality evidence that physical activity and exercise improved cognitive performance measures and patient-reported measures of cognitive function in cancer survivors.
Childhood cancer survivors may be at risk for cognitive decline throughout their lives (even if not present in the first 10 years after therapy). In a study of 2,375 adult survivors of childhood ALL, Hodgkin lymphoma, or CNS tumors (mean age at evaluation, 31.8 years) and their sibling controls, new onset memory impairment emerged more often in survivors, decades after cancer diagnosis and treatment.[15]
Compared with siblings, a higher proportion of survivors with no impairment in memory at baseline had new-onset memory impairment at follow-up, as follows:
Sibling proportion, 7.8%.
ALL survivors treated with chemotherapy only, 14.0%.
ALL survivors treated with cranial radiation therapy, 25.8%.
CNS tumor survivors, 34.7%.
Hodgkin lymphoma survivors, 16.6%.
Factors associated with new-onset memory impairment included cranial radiation in survivors of CNS tumor (relative risk [RR], 1.97) and dose of 8,000 mg/m2 or more of alkylator chemotherapy in survivors of ALL treated without cranial radiation therapy (RR, 2.80).
Neurological conditions mediated the impact of cranial radiation therapy on new-onset memory impairment in survivors of CNS tumors.
Smoking, obesity, low education attainment, and low physical activity were associated with an elevated risk of new onset-memory impairment.
Neurocognitive outcomes in brain tumor survivors
Long-term cognitive effects caused by illness and associated treatments are well-established morbidities in survivors of childhood and adolescent brain tumors. Risk factors for adverse neurocognitive effects in this group include the following:
Cranial radiation therapy.
Cranial radiation therapy has been associated with the highest risk of long-term cognitive morbidity, particularly in younger children.[3,4]
There is an established dose-response relationship, with patients who receive higher-dose cranial radiation therapy consistently performing more poorly on intellectual measures.[3,4,16]
Radiation dose to specific regions of the brain, including the temporal lobes and hippocampi, have been shown to significantly impact longitudinal IQ scores and academic achievement scores among children treated with craniospinal irradiation for medulloblastoma.[17]
The negative impact of radiation treatment has been characterized by changes in IQ scores, which have been noted to drop about 2 to 5 years after diagnosis.[24–26]
The decline continues 5 to 10 years afterward, although less is known about potential stabilization or further decline of IQ scores several decades after diagnosis.[24,26]
The decline in IQ scores over time typically reflects the child’s failure to acquire new abilities or information at a rate similar to that of his or her peers, rather than a progressive loss of skills and knowledge.[27]
Affected children also may experience deficits in other cognitive areas, including academic domains (reading and math) and problems with attention, processing speed, memory, and visual or perceptual motor skills.[28]
Changes in cognitive functioning may be partially explained by radiation-induced reduction of normal-appearing white matter volume or integrity of white matter pathways, as evaluated through magnetic resonance imaging (MRI).[29,30]
Reduced white matter integrity has been directly linked to slowed cognitive processing speed in survivors of brain tumors,[31] while greater white matter volume has been associated with better working memory, particularly in females.[30]
Data from contemporary protocols show that using lower doses of cranial radiation, proton beam radiation, and more targeted treatment volumes appears to reduce the severity of neurocognitive effects of therapy.[3,19]
Evidence (predictors of cognitive decline among survivors of CNS tumors):
Longitudinal cohort studies have provided insight into the trajectory and predictors of cognitive decline among survivors of CNS tumors.
A multisite, prospective, longitudinal trial evaluated predictors of cognitive performance among 139 infants with brain tumors who were treated with chemotherapy, with or without focal proton or photon radiation therapy.[32]
IQ, parent-reported working memory, and parent-reported adaptive functioning were worse than normative expectations at baseline, with younger age and lower SES representing predictive factors.
IQ remained stable over time, whereas parent-reported attention and executive dysfunction increased.
Cognitive outcomes did not differ by treatment exposure (chemotherapy only vs. chemotherapy with radiation therapy).
Changes in cognitive function were associated with supratentorial tumor location and cerebrospinal fluid diversion.
St. Jude Children’s Research Hospital (SJCRH) studied 78 children younger than 20 years (mean, 9.7 years) diagnosed with a low-grade glioma.[33]
Cognitive decline after 54 Gy of conformal cranial radiation therapy was noted (see Figure 5).
Age at time of cranial irradiation was more important than was cranial radiation dose in predicting cognitive decline, with children younger than 5 years estimated to experience the greatest cognitive decline.
In a study of 51 children with low-grade gliomas and low-grade glioneural tumors diagnosed within the first year of life, the following was reported:[34]
Mean IQ score was 75.5; 75% of the children had IQ scores lower than 85.
Predictors of low IQ included a supratentorial location of the primary tumor and treatment with more chemotherapy regimens but not radiation use.
The child’s ability to complete age-appropriate tasks was as affected as IQ scores.
A study of 126 medulloblastoma survivors treated with 23.4 Gy or 36 Gy to 39.6 Gy of craniospinal radiation (with a conformal boost dose of 55.8 Gy to the primary tumor bed) assessed processing speed, attention, and memory performance.[35]
Processing speed scores declined significantly over time, while less decline was observed in attention and memory performance. Higher doses of radiation and younger age at diagnosis predicted slower processing speed over time.
Studies of working memory and academic achievement in patients enrolled on the same medulloblastoma trial (SJCRH SJMB03 [NCT00085202]) indicated that performance was largely within the age-expected range up to 5 years postdiagnosis,[36,37] although in both studies, posterior fossa syndrome, higher cranial radiation dose, and younger age at diagnosis predicted worse performance over time.
Serious hearing loss was associated with intellectual and academic decline over time.[37]
In a prospective study of 178 children with medulloblastoma, 60 (34%) developed posterior fossa syndrome. Of these patients, 40 (23%) developed complete mutism and 20 (11%) developed diminished speech.[38]
All children with posterior fossa syndrome had severe ataxia, and 42.5% of patients with posterior fossa syndrome and complete mutism had movement disorders.
Independent risk factors for posterior fossa syndrome included younger age and surgery in a low-volume surgery center. Risk was lower among children with sonic hedgehog (SHH) tumors.
Speech and gait returned in children with posterior fossa syndrome and complete mutism at a median of 2.3 and 0.7 months, respectively. Speech and gait returned in children with posterior fossa syndrome and diminished speech at a median of 2.1 and 1.5 months, respectively. However, with 12 months of follow-up, 12 of 27 children (44.4%) with posterior fossa syndrome and complete mutism were nonambulatory at 1 year.
The presence of a movement disorder or high ataxia score was associated with delayed speech recovery, whereas older age and high ataxia score were associated with delayed gait return.
Symptoms improved in all children, but no child with posterior fossa syndrome had a normal neurological examination at a median of 23 months after surgery.
A prospective study compared 36 pediatric patients with medulloblastoma who experienced posterior fossa syndrome with 36 patients with medulloblastoma who did not experience posterior fossa syndrome but were matched on treatment and age at diagnosis.[38,39]
The posterior fossa syndrome group demonstrated lower mean scores at 1, 3, and 5 years postdiagnosis on general intellectual ability, processing speed, working memory, and spatial relations compared with the non–posterior fossa syndrome group.
The group who experienced posterior fossa syndrome showed little recovery over time and further decline over time in some domains (attention and working memory), compared with the non–posterior fossa syndrome group.
Canadian investigators evaluated the impact of radiation (dose and boost volume) and neurological complications on patterns of intellectual functioning in a cohort of 113 medulloblastoma survivors (mean age at diagnosis, 7.5 years; mean time from diagnosis to last assessment, 6 years).[4]
Survivors treated with reduced-dose craniospinal radiation therapy plus tumor bed boost showed stable intellectual functioning.
Neurological complications, such as hydrocephalus requiring cerebrospinal fluid diversion and mutism, and treatment with higher doses and larger boost volumes of radiation resulted in intellectual declines with distinctive trajectories.
Studies are beginning to examine cognitive outcomes in histologically distinct subtypes of brain tumors.
Data from a sample of 121 medulloblastoma patients demonstrated variation in cognitive outcomes by four distinct molecular subgroups and differences in patterns of change over time.[40]
Future research is required to establish if neurocognitive outcomes vary across biologically distinct subtypes of childhood brain tumors.
SES has been shown to predict long-term cognitive outcomes in brain tumor survivors. In a prospective, longitudinal, phase II study of 248 children who were treated with conformal radiation therapy (54–59.4 Gy) for ependymoma, low-grade glioma, or craniopharyngioma, patients were monitored serially with cognitive assessment for 10 years.[23]
At preradiation therapy baseline, significant associations were seen between SES and IQ, reading and math scores, sustained attention, and adaptive function. Higher SES was associated with better performance (P < .005).
SES predicted change over time in IQ and reading and math scores. Higher SES was associated with less decline (P < .001).
These results suggest that higher SES is a protective factor for cognitive late effects.
Evidence (predictors of cognitive decline among long-term survivors of CNS tumors):
Although adverse neurocognitive outcomes observed 5 to 10 years after treatment are presumed to be pervasive, and potentially worsen over time, few empirical data are available regarding the neurocognitive functioning in very long-term survivors of CNS tumors.
A longitudinal study evaluated the relationship of hippocampal dose and short-term memory decline in 80 children and adolescents with low-grade gliomas (median age, 9.5 years at treatment) who received 54 Gy of radiation therapy.[41]
At a median neurocognitive follow-up of 9.8 years, higher hippocampal dose (volume receiving 40 Gy) was associated with a greater decline in delayed recall.
Among adult survivors participating in the CCSS, CNS tumor survivors (n = 802) self-reported significantly more problems with attention/processing speed, memory, emotional control, and organization than did survivors of non-CNS malignancies (n = 5,937) and sibling controls (n = 382).[42]
Another CCSS study evaluating patterns of late mortality and morbidity in 2,821 adult survivors of CNS tumors reported impairment on measures of attention/processing speed (42.9%–73.3%) and memory (14.3%–37.4%), with differences observed by diagnosis and cranial radiation dose.[43]
A study of 224 adult survivors of pediatric brain tumors participating in the St. Jude Life (SJLIFE) cohort study revealed that 20% to 30% of the survivors demonstrated severe neurocognitive impairment (defined as at least two standard deviations below normative mean) on clinical assessments of intelligence, memory, and executive function (e.g., planning, organization, and flexibility).[3]
Among adults in the general population, the expected impairment rate at this threshold is 2%.
Survivors who received whole-brain cranial irradiation were 1.5 to 3 times more likely to have severe neurocognitive impairment than were survivors who did not receive any cranial irradiation.
Hydrocephalus with shunt placement and seizures were also associated with increased risk of impairment.
In the CCSS, investigators compared long-term neuropsychological and SES outcomes of 181 adult survivors of pediatric low-grade gliomas with the outcomes of an age-matched and sex-matched sibling comparison group.[44]
Survivors who were treated with surgery and radiation therapy (median age at diagnosis, 7 years; median age at assessment, 41 years) scored lower on estimated IQ than did survivors who were treated with surgery only, who scored lower than siblings (surgery and radiation therapy, 93.9; surgery only, 101.2; siblings, 108.5; all P values < .0001).
Younger age at diagnosis was predictive of low scores for all neuropsychological outcomes except for attention/processing speed.
Survivors who were treated with surgery and radiation therapy had more-than-twofold–lower occupation scores, income, and education than did survivors who were treated with surgery only.
In a retrospective review of 528 brain tumor survivors diagnosed between 2000 and 2015, the prevalence of a clinical diagnosis of ADHD was 13.1%.[45]
Of the survivors, 12.1% used medications for ADHD, and 19.9% of survivors had symptoms of ADHD without a clinical diagnosis.
ADHD diagnosis was associated with younger age at tumor diagnosis and supratentorial tumor location, but not with sex, tumor type, or treatment type.
The neurocognitive consequences of CNS disease and treatment may have a considerable impact on functional outcomes for brain tumor survivors.
In childhood and adolescence, neurocognitive deficits have been associated with poor social adjustment, including problems with peer relations, social withdrawal, and reduced social skills.[46,47]
CNS tumor survivors are more likely to need special education services than are survivors of other malignancies.[48]
Adult CNS tumor survivors are less likely to live independently, marry, and graduate from college than are survivors of other malignancies and siblings.[49]
Cognitive outcomes after proton radiation therapy
Data are emerging regarding cognitive outcomes after proton radiation to the CNS.[50–52] However, these studies have been limited by retrospective analysis of cognitive outcomes among relatively small clinically heterogenous pediatric brain tumor cohorts and the use of historically treated photon patients or population standards as comparison groups.
In studies largely describing IQ changes during early follow-up (<5 years from radiation), results demonstrate lack of difference in slopes of IQ change among photon-treated and proton-treated patients [50] and significant declines in cognitive processing speed among patients treated with proton radiation.[51]
One study compared the intellectual trajectories between pediatric patients with medulloblastoma who were treated with proton and photon radiation therapy (4.3-year mean follow-up after median 23.4-Gy craniospinal irradiation dose). Notably, boost dose and margins were significantly different between the two groups.[52]
Children treated with proton radiation therapy exhibited superior long-term outcomes in global IQ, perceptual reasoning, and working memory compared with children who were treated with photon radiation therapy.
The photon radiation therapy group exhibited a significant decline in global IQ, working memory, and processing speed.
The proton radiation therapy group exhibited stable scores over time in all domains, except for processing speed.
Considering the relatively brief follow-up time from radiation, longitudinal follow-up is important to determine whether proton radiation provides a clinically meaningful benefit in sparing cognitive function compared with photon radiation. In addition, more targeted radiation treatment volumes with photons may diminish potential differences.
Neurocognitive outcomes in acute lymphoblastic leukemia (ALL) survivors
To minimize the risk of late cognitive sequelae, contemporary therapy for ALL uses a risk-stratified approach that reserves cranial irradiation for children who are considered at high risk of CNS relapse.
ALL and cranial radiation
In survivors of ALL, cranial radiation therapy may result in clinical and radiographic neurological late sequelae, including the following:
Clinical leukoencephalopathy. Clinical leukoencephalopathy characterized by fluctuating encephalopathy, stroke-like symptoms, spasticity, ataxia, dysarthria, dysphagia, hemiparesis, and seizures is uncommon after contemporary ALL therapy. Neuroimaging frequently demonstrates white matter abnormalities among survivors treated with cranial irradiation and/or high-dose methotrexate.
Radiographic leukoencephalopathy has been reported in up to 80% of children on some treatment regimens.
Higher doses and more courses of intravenous methotrexate have been reported to increase risk of leukoencephalopathy.[53]
In many patients, white matter anomalies are transient and decrease in prevalence, extent, and intensity with longer elapsed time from completion of therapy.[53]
Leukoencephalopathy results in smaller white matter volumes that have been correlated with cognitive deficits.[53]
Although these abnormalities are mild among the irradiated patients (overall IQ decline of approximately 10 points), those who have received higher doses at a young age may have significant learning difficulties.[54,55]
Neurocognitive deficits. Deficits in neurocognitive functions such as visual-motor integration, processing speed, attention, and short-term memory are reported in children treated with 18 Gy to 24 Gy.[54,56,57]
Females and children treated at a younger age are more vulnerable to the adverse impact of cranial radiation on the developing brain.[5]
The decline in intellectual functioning appears to be progressive, showing more impairment of cognitive function with increasing time since radiation therapy.[5,6]
Limited studies suggest that long-term survivors of childhood ALL treated with cranial irradiation are at risk of progressive decline consistent with early-onset mild cognitive impairment; this risk is most prominent among those treated with cranial radiation doses of 24 Gy.[58,59]
ALL and chemotherapy-only CNS therapy
Because of its penetrance into the CNS, systemic methotrexate has been used in a variety of low-dose and high-dose regimens for leukemia CNS prophylaxis.
Systemic methotrexate in high doses with or without radiation therapy can lead to an infrequent but well-described leukoencephalopathy, which has been linked to neurocognitive impairment.[53]
When neurocognitive outcomes after radiation therapy and chemotherapy-only regimens are directly compared, the evidence suggests a better outcome for those treated with chemotherapy alone, although some studies show no significant difference.[60,61]
In a longitudinal analysis of 210 childhood ALL survivors, the development of acute leukoencephalopathy during chemotherapy-only CNS therapy predicted higher risks of developing long-term neurobehavioral problems (e.g., deficits in organization and task initiation [components of executive function]) and reduced white matter integrity in frontal brain regions.[62]
Compared with cranial irradiation, chemotherapy-only CNS-directed treatment produces neurocognitive deficits involving processes of attention, speed of information processing, memory, verbal comprehension, visual-spatial skills, visual-motor functioning, and executive functioning. Global intellectual function is typically preserved.[56,60,63–65]
Few longitudinal studies evaluating long-term neurocognitive outcome report adequate data for a decline in global IQ after treatment with chemotherapy alone.[63]
The academic achievement of ALL survivors in the long term seems to be generally average for reading and spelling, with deficits mainly affecting arithmetic performance.[60,66]
Risk factors for poor neurocognitive outcome after chemotherapy-only CNS-directed treatment are younger age and female sex.[65,67]
Reduced cognitive status has been observed in association with reduced integrity in neuroanatomical regions essential in memory formation (e.g., reduced hippocampal volume with increased activation and thinner parietal cortices).[62]
The long-term impact of these prevalent neurocognitive and neuroimaging abnormalities on functional status in aging adults treated for childhood ALL, particularly those treated with contemporary approaches using chemotherapy alone, remains an active area of research.
Research in childhood ALL survivors treated with chemotherapy alone suggests that genetic predisposition may modulate risk of developing neurocognitive late effects. It also provides insights into potential mechanisms underlying neurocognitive deficits.[68,69]
Evidence (neurocognitive functioning in large pediatric cancer survivor cohorts):
CCSS investigators identified sex-specific associations between treatment and chronic health conditions with neurocognitive impairment among 1,207 survivors of ALL (mean age, 30.6 years) who were treated with chemotherapy only.[7]
Both male and female survivors reported increased prevalence of impaired task efficiency (attention and processing speed; adjusted odds ratio [OR], 1.89 for males; OR, 1.50 for females) and impaired memory (OR, 1.89 for males; OR, 1.96 for females) compared with 2,273 same-sex siblings.
Among male survivors, impaired task efficiency was associated with grades 2 to 4 neurological conditions (OR, 4.33) and pulmonary conditions (OR, 4.99). Impaired memory was associated with increased cumulative dose of intrathecal methotrexate (OR, 1.68) and exposure to dexamethasone (OR, 2.44).
In female survivors, grades 2 to 4 endocrine conditions were associated with higher risk of impaired task efficiency (OR, 2.19) and memory (OR, 2.26).
The CCSS examined parent-reported cognitive, behavior, and learning problems from 1,560 adolescent survivors of childhood ALL who were treated with chemotherapy alone between 1970 and 1999.[70]
Survivors treated with cranial irradiation had significantly higher frequency of problems in anxiety-depression, inattention-hyperactivity, and social withdrawal than did patients who were not treated with cranial irradiation.
Compared with siblings, survivors treated with chemotherapy only were more likely to demonstrate headstrong behavior (19% of survivors vs. 14% of siblings, P = .010), inattention-hyperactivity (19% vs. 14%, P < .0001), social withdrawal (18% vs. 12%, P = .002), and had higher rates of learning problems (28% vs. 14%, P < .0001).
In multivariable models among survivors, increased cumulative dose of intravenous methotrexate (i.e., >4.3 g/m2) conferred increased risk of inattention-hyperactivity (RR, 1.53).
Adolescent survivors with cognitive or behavior problems and those with learning problems were less likely to graduate from college as young adults than adolescent survivors without cognitive or behavior problems.
Inattention and hyperactivity problems were associated with the highest risk of special education placement during adolescence. Participation in special education during adolescence did not improve adult educational attainment.
The SJCRH TOTAL XVI (NCT00549848) trial evaluated whether a higher dose of pegaspargase and early intensification of triple intrathecal therapy (ITT; methotrexate, hydrocortisone, and cytarabine) would improve systemic and CNS control in children with newly diagnosed ALL. Intensified ITT was defined as more than 21 cumulative doses for patients with low-risk ALL (70 of 194) and more than 27 doses for those with standard- and high-risk ALL (81 of 206). Neurocognitive testing was completed at the end of therapy.[71]
The overall group had below age-based norms on measures of global intelligence (P < .0001), attention (P = .0051), working memory (P = .0001), processing speed (P = .0002), fine motor speed (P = .0001), and math skills (P = .0087). Caregiver ratings of patient functioning showed elevated risks of problems in attention (P = .0173), executive function (P = .0001), and adaptive skills (P = .0001).
In the low-risk group, there were no significant differences between patients treated with and without intensified ITT.
Patients with standard- and high-risk ALL who were treated with intensified ITT had poorer working memory (P = .0328) and fine motor speed (P = .0403), and elevated ratings of inattention (P = .0189) and executive dysfunction (P = .0245). Females in this risk group treated with intensified ITT had lower working memory scores.
In the SJCRH Total XV (NCT00137111) trial, which omitted prophylactic cranial irradiation, comprehensive cognitive testing of 243 participants at week 120 revealed the following:[72]
A higher risk of below-average performance on a measure of sustained attention but not on measures of intellectual functioning, academic skills, or memory.
The risk of cognitive deficits correlated with treatment intensity but not with age at diagnosis or sex.
Prolonged follow-up (average, 7.7 years from diagnosis) of this cohort demonstrated that intelligence was within normal limits compared with population expectations, but measures of executive function, processing speed, and memory were less than population means.
Higher plasma methotrexate was associated with executive dysfunction, thicker cerebral cortex, and higher activity in frontal brain regions on functional MRI.
In a large prospective study of neurocognitive outcomes in children with newly diagnosed ALL, 555 children were randomly assigned to receive CNS-directed therapy according to risk group.[73]
The low-risk group was randomly assigned to receive either intrathecal methotrexate or high-dose methotrexate.
The high-risk group was randomly assigned to receive either high-dose methotrexate or 24 Gy of cranial radiation therapy.
A significant reduction in IQ scores (4–7 points) was observed in all patient groups when compared with controls, regardless of the CNS treatment delivered.
Children younger than 5 years at diagnosis were more likely to have IQs below 80 at 3 years posttherapy than were children older than 5 years at diagnosis, irrespective of treatment allocation, suggesting that younger children are more vulnerable to treatment-related neurological toxic effects.
Persistent cognitive deficits and progressive intellectual decline have been observed in cohorts of adults treated for ALL during childhood and associated with reduced educational attainment and unemployment.[5,55,59] The results of a study of more than 500 adult survivors of childhood ALL (average, 26 years postdiagnosis) showed the following:[55]
Survivors demonstrated increased rates of impairment across all neurocognitive domains (ranging from 28.6%–58.9% for each domain).
Rate of severe impairment increased as a function of cranial radiation dose and was common among survivors treated with lower doses of cranial irradiation and chemotherapy only.
Impairment in executive function skills increased with time since diagnosis in a cranial radiation dose-dependent manner; impairment in intellect, academics, and memory progressively increased with younger age at treatment in a cranial radiation dose-dependent manner; and neurocognitive impairment was related to functional outcomes as adults, including reduced likelihood of college graduation and full-time employment.
ALL and steroid therapy
The type of steroid used for ALL systemic treatment may affect cognitive functioning.
In a study that involved long-term neurocognitive testing (mean follow-up, 9.8 years) of 92 children with a history of standard-risk ALL who had received either dexamethasone or prednisone during treatment, no meaningful differences in mean neurocognitive and academic performance scores were observed.[74]
In contrast, in a study of 567 adult survivors of childhood leukemia (mean age, 33 years; mean time since diagnosis, 26 years), the following was reported:[55]
Dexamethasone exposure was associated with increased risk of impairment in attention (RR, 2.12; 95% confidence interval [CI], 1.11–4.03) and executive function (RR, 2.42; 95% CI, 1.20–4.91), independent of methotrexate exposure.
Intrathecal hydrocortisone also increased risk of attention problems (RR, 1.24; 95% CI, 1.05–1.46).
Other cancers
Neurocognitive abnormalities have been reported in other groups of cancer survivors. A study of adult survivors of childhood non-CNS cancers (including ALL, n = 5,937) reported the following:[57]
13% to 21% of survivors reported impairment in task efficiency, organization, memory, or emotional regulation. This rate of impairment was approximately 50% higher than that reported in the sibling comparison group.
Factors such as diagnosis before age 6 years, female sex, cranial radiation therapy, and hearing impediment were associated with impairment.
Emerging data suggest that the development of chronic health conditions in adulthood may contribute to cognitive deficits in long-term survivors of non-CNS cancers.
An SJLIFE cohort study evaluated whether children who experienced CNS injury were at higher risk of neurocognitive impairment associated with subsequent late-onset chronic health conditions. A total of 2,859 survivors who were aged 18 years or older and at least 10 years from diagnosis completed a neurocognitive battery and clinical examination. Of these patients, 1,598 had received CNS-directed therapy, including cranial radiation, intrathecal methotrexate, or neurosurgery.[75]
CNS-treated survivors performed worse than non–CNS-treated survivors on all neurocognitive tests, and they were more likely to have global neurocognitive impairment (46.9% vs. 35.3%).
A dose-response association was observed between severity/burden score of chronic health conditions and global neurocognitive impairment only among CNS-treated survivors (high OR, 2.24; 95% CI, 1.42–3.53; very high OR, 4.07; 95% CI, 2.30–7.17).
Cardiovascular and pulmonary conditions were associated with cognitive outcomes (processing speed, executive function, and memory impairment) only in CNS-treated survivors with neurological conditions.
Neurocognitive abnormalities have been reported for the following cancers:
Acute myeloid leukemia (AML). CCSS investigators compared the neurocognitive and psychosocial outcomes of 482 survivors (median age, 30 years) with those of 3,190 sibling controls (median age, 32 years).[76]
Compared with siblings, AML survivors were more likely to report impairment in overall emotional (RR, 2.19; 95% CI, 1.51–3.18), neurocognitive (RR, 2.03; 95% CI, 1.47–2.79), and physical quality of life (RR, 2.71; 95% CI, 1.61–4.56) outcomes.
Survivors were also at increased risk of psychosocial deficits, including lower educational attainment (RR, 1.15; 95% CI, 1.03–1.30), unemployment (RR, 1.41; 95% CI, 1.16–1.71), and lower income (RR, 1.39; 95% CI, 1.17–1.65).
Outcomes were not statistically different between survivors who were treated with transplant and survivors who were treated with intensive chemotherapy alone.
Osteosarcoma. In a study evaluating neurocognitive function among 80 long-term survivors of osteosarcoma (mean time since diagnosis, 24.7 years), survivors demonstrated lower mean scores in reading skills, attention, memory, and processing speed than did community controls.[77]
The presence of cardiac, pulmonary, and endocrine conditions were significantly associated with worse performance on measures of memory and processing speed.
Soft tissue sarcoma. A CCSS report characterized the prevalence of and risk factors for self-reported neurocognitive impairment in survivors of rhabdomyosarcoma (n = 713) and their siblings (n = 706). The report also identified risk factors associated with these outcomes in survivors.[78]
Compared with their siblings, more survivors reported neurocognitive impairments (task efficiency: 21.1% vs. 13.7%; emotional regulation: 16.7% vs. 11.0%; memory: 19.3% vs. 15.1%). Cranial radiation exposure increased the risk of impaired task efficiency (OR, 2.30).
Smoking was associated with impaired task efficiency (OR, 2.06) and memory (OR, 2.23).
Neurological and hearing conditions were associated with impaired memory (OR, 2.44 and OR, 1.87, respectively).
SJLIFE study investigators evaluated neurocognitive function and health status through objective clinical assessments in 150 survivors of childhood soft tissue sarcoma (median age, 33 years; median time from diagnosis, 24 years).[79]
Compared with community and population controls, survivors demonstrated lower measures of verbal reasoning, mathematics, and long-term memory.
Cumulative anthracycline exposure (per 100 mg/m2) was found to be associated with poorer verbal reasoning, reading, and patient-reported vitality.
Neurological and neurosensory chronic conditions were associated with poorer mathematics scores and hearing impairment.
Better cognitive performance was associated with higher social attainment.
Wilms tumor. SJLIFE study investigators examined the prevalence and predictors of neurocognitive outcomes, social attainment, emotional distress, and health-related quality of life in long-term survivors of pediatric Wilms tumor (n = 158; median time since diagnosis, 29 years; median age, 33 years) and compared the results to community controls.[80]
Long-term Wilms tumor survivors demonstrated lower neurocognitive function, including verbal reasoning, academics, attention, memory, and executive function, than did controls.
Wilms tumor survivors were less likely to graduate from college (OR, 2.23) and had more moderate-to-severe neurological conditions than did controls (18.4% vs. 8.2%; P < .001).
Retinoblastoma. Serial assessment of cognitive and adaptive functioning in a group of retinoblastoma survivors younger than 6 years revealed declines in developmental functioning over time, with the most pronounced declines observed in patients with 13q deletions.[81]
Further longitudinal follow-up of this cohort identified improvement in adaptive functioning in all treatment groups and in cognitive function in survivors who were treated with enucleation alone from the age of 5 years to the age of 10 years.[82]
At age 10 years, overall functioning was generally within the average range, although estimated IQ was significantly below the normative mean for children who were treated with enucleation alone.[82]
A study of very long-term adult survivors, who were on average 33 years postdiagnosis, demonstrated largely average cognitive functioning across domains of intelligence, memory, attention, and executive function.[83]
Lymphoma. Survivors of lymphoma have not historically been considered at risk of developing neurocognitive late effects.
However, one report observed that more than two-thirds of survivors of childhood non-Hodgkin lymphoma experienced at least mild neurocognitive impairment, including severe deficits in executive function (13%), attention (9%), and memory (4%).[84]
Similarly, in a study of 62 adult survivors of childhood Hodgkin lymphoma, survivors demonstrated worse performance on measures of sustained attention, short- and long-term memory, and cognitive fluency when compared with national normative data.[85] Importantly, measures of cardiac and pulmonary function were also associated with neurocognitive impairment in this group of survivors.
Neuroblastoma. CCSS investigators compared cognitive outcomes among 837 survivors of neuroblastoma (median age, 1 year at diagnosis; median attained age, 25 years) and a sibling cohort (median attained age, 23 years) using the CCSS Neurocognitive Questionnaire (NCQ).[86]
Survivors had a 50% higher risk of impairment with attention/processing speed and emotional regulation.
The risk of neurocognitive impairments was higher among survivors with chronic health conditions.
Survivors were also less likely to attain adult milestones, such as living independently.
Hematopoietic stem cell transplant (HSCT)
Cognitive and academic consequences of HSCT in children have also been evaluated and include, but are not limited to, the following:
A survey study evaluated late neurocognitive outcomes among 199 survivors of HSCT (HSCT at age <21 years; median follow-up from HSCT, 27.6 years).[87]
As assessed by the CCSS NCQ, 18.9% to 32.5% of survivors reported impairments in task efficiency, memory, emotional regulation, or organization, compared with the expected rate of 10% (general population).
Quality of life, as assessed by Neuro-Quality of Life Cognitive Function Short Form (Neuro-QoL), among survivors (average T score, 49.6) was comparable to that of the normative population (50).
Independent risk factors for impaired Neuro-QoL (T score <40) included hearing issues (OR, 4.7; 95% CI, 1.96–12.6), history of stroke or seizure (OR, 4.46; 95% CI, 1.44–13.8), and sleep disturbances (OR, 6.96; 95% CI, 2.53–19.1).
SJCRH investigators examined the influence of age and conditioning with total-body irradiation (TBI) on the trajectory of cognitive function after HSCT. Among 315 patients who completed a baseline assessment, 183 were alive at 1 year after HSCT and completed additional assessments at 1, 3, and 5 years.[88]
At 5 years after HSCT, younger patients (aged <3 years at baseline) who received TBI demonstrated a significantly lower IQ than those who did not receive TBI.
Longitudinal analyses demonstrated a significant impact of age and TBI over time with declines in cognitive functioning during the first year among the youngest patients and recovery of functioning in subsequent years among patients who did not receive TBI.
Young patients who received TBI failed to recover the losses experienced during the first year after HSCT, demonstrating stability in their functioning, but at a lower level.
Neurological Sequelae
Risk of neurological complications may be predisposed by the following:
Tumor location.
Neurosurgical procedures.
Cranial radiation therapy.
Spinal radiation therapy.
Specific neurotoxic chemotherapeutic agents.
In children with CNS tumors, mass effect, tumor infiltration, and increased intracranial pressure may result in motor or sensory deficits, cerebellar dysfunction, and secondary effects such as seizures and cerebrovascular complications.[89]
Numerous reports describe abnormalities of CNS integrity and function, but such studies are typically limited by small sample size, cohort selection and participation bias, cross-sectional ascertainment of outcomes, and variable time of assessment from treatment exposures. In contrast, relatively few studies comprehensively or systematically ascertain outcomes related to peripheral nervous system function.
CNS tumor survivors remain at higher risk of new-onset adverse neurological events across their lifetimes than siblings. No plateau has been reached for new adverse sequelae, even 30 years from diagnosis, according to a longitudinal study of 1,876 5-year survivors of CNS tumors from the CCSS. The median time from diagnosis was 23 years, and the median age of the patients studied was 30.3 years.[90]
Cranial radiation, stroke, tumor recurrence, and development of meningioma were independently associated with late-onset neurological sequelae (seizures, focal neurological dysfunction, and neurosensory abnormalities).
This finding supports the need to monitor these patients carefully with continued neurological follow-up within or in close association with a multidisciplinary cancer survivor clinic.
Neurological complications that may occur in survivors of childhood cancer include the following:
Seizures
The development of seizures may occur secondary to tumor mass effect within the CNS and/or from neurotoxic CNS-directed therapies.
In 1,876 5-year survivors of CNS tumors from the CCSS, the incidence of seizures increased from 27% in survivors 5 years from diagnosis to 41% in survivors 30 years from diagnosis.[90]
Late-onset seizures were associated with frontal lobe radiation of 50 Gy (hazard ratio [HR], 1.8) and temporal lobe radiation in a dose-dependent fashion (HR, 1.9 for 1–49 Gy; HR, 2.2 for >50 Gy).
Other risk factors associated with late-onset seizures included recurrence (HR, 2.3), development of meningioma (HR, 2.6), and history of stroke (HR, 2.0).
The risk of seizures was elevated for survivors compared with siblings (HR, 12.7).
Among survivors of childhood leukemia in the CCSS (N = 4,151; 64.5% treated with cranial irradiation), 6.1% reported the development of a seizure disorder, and seizures occurred more than 5 years after diagnosis in 51% of these patients.[91]
An SJLIFE study evaluated the impact of seizure-related factors on neurocognitive, health-related quality of life (HRQOL), and social outcomes in 2,022 childhood cancer survivors (median age, 31.5 years; median time from diagnosis, 23.6 years).[92]
Seizures were identified in 232 survivors (11.5%; 29.9% of survivors with CNS tumors and 9.0% of survivors without CNS tumors).
Among CNS tumor survivors, seizures were associated with poorer (based on expected age-adjusted standard z-scores) executive function and processing speed. Seizure resolution was associated with improved attention and memory.
Among non-CNS tumor survivors, seizures were associated with worse function in all cognitive domains compared with survivors without seizures. Seizure severity was associated with worse processing speed, and resolution of seizures was associated with better executive function and attention.
Non-CNS survivors with seizures had poorer HRQOL outcomes (physical functioning, physical role limitations, and general health). Seizures in both non-CNS and CNS tumor survivors increased the risk of unemployment and part-time employment.
Peripheral neuropathy
Vinca alkaloid agents (vincristine and vinblastine) and heavy metals (cisplatin and carboplatin) may cause peripheral neuropathy.[93–95]
Peripheral neuropathy presents during treatment and appears to improve or clinically resolve after completion of therapy.[93] However, recent studies of long-term survivors suggest that chemotherapy-induced peripheral neuropathy is likely under-ascertained.[96]
Higher cumulative doses of vincristine and/or intrathecal methotrexate have been linked to neuromuscular impairments in long-term survivors of childhood ALL, which suggests that persistent effects of these agents may affect functional status in aging survivors.[93]
SJLIFE study investigators observed associations between peripheral neuropathy and impairment in performance measures of movement (mobility and walking endurance) and quality of life (physical functioning, role physical, and general health) among survivors of childhood ALL.[97]
Static-standing balance impairment (a predictor of falling within the next 90 days) was more common in survivors compared with controls but was not associated with peripheral neuropathy.[97]
Among adult survivors of extracranial solid tumors of childhood (median time from diagnosis, 25 years), standardized assessment of neuromuscular function disclosed motor impairment in association with vincristine exposure and sensory impairment in association with cisplatin exposure.[94]
Survivors with sensory impairment demonstrated a higher prevalence of functional performance limitations related to poor endurance and mobility restrictions.
Stroke or other cerebrovascular effects
Childhood CNS tumor survivors have a 43-fold elevated risk of stroke compared with siblings.[43,98]
Cranial radiation therapy (dose dependent), baseline atherosclerosis, hypertension, and African American ethnicity are identified risk factors.[99–101] For information about stroke, see the Cerebrovascular disease section.
A British CCSS (n = 13,457 survivors) that included 2,885 childhood CNS tumor survivors reported the following:[102]
CNS tumor survivors who were treated with cranial irradiation had an increased risk of developing cerebrovascular disease between the ages of 50 years and 65 years.
Compared with an expected incidence of 4.2%, the cumulative incidence of cerebrovascular disease in survivors was 11.6% by age 40 years, 16% by age 50 years, and 26% by age 65 years.
Among 271 CCSS participants with a history of stroke at a median age of 19 years, 70 reported a second stroke at a median age of 32 years.[103]
The 10-year cumulative incidence of late recurrent stroke was 21% overall, and 33% for those treated with 50 Gy or higher cranial radiation therapy.
Risk factors for recurrent stroke included the following:
Older age at first stroke (HR, 6.4; 95% CI, 1.8–23).
A PENTEC review identified 101 of 3,989 pediatric patients who experienced at least one cerebrovascular toxicity (transient ischemic attack, stroke, moyamoya, or arteriopathy).[104]
Predicted incidences were based on the radiation dose to the Circle of Willis: 0.2% at 30 Gy, 1.3% at 45 Gy, and 4.4% at 54 Gy.
At an attained age of 35 years, the predicted stroke incidence was 0.9% to 1.3% for 30 Gy, 1.8% to 2.7% for 45 Gy, and 2.8% to 4.1% for 54 Gy (compared with a baseline risk of 0.2%–1.0%).
At an attained age of 45 years, the predicted stroke incidence was 1.2% to 4.2% for 30 Gy, 4.5% to 8.6% for 45 Gy, and 6.7% to 13% for 54 Gy (compared with a baseline risk of 0.5%–1.0%).
Hypersomnia (daytime sleepiness) or narcolepsy
In a retrospective review of brain tumor patients treated at SJCRH, investigators identified 39 of 2,336 patients who were diagnosed with hypersomnia/narcolepsy, for a prevalence rate of 1,670 cases per 100,000, which is much higher than a prevalence rate of 20 to 50 cases per 100,000 reported in the general population.[105]
This may be an underestimate in childhood brain tumor survivors because many patients with mild-to-moderate symptoms, such as fatigue and sleep disturbances, may not be recognized or referred to a sleep specialist.
Hypersomnia/narcolepsy was diagnosed at a median of 6 years (range, 0.4–13.2 years) from tumor diagnosis and 4.7 years (range, 1.5–10.4 years) from cranial radiation.
Midline tumor location and antiepilepsy drug use correlated with hypersomnia/narcolepsy, while radiation dose higher than 30 Gy trended toward significance.
Posterior fossa tumor location was associated with a reduced risk of hypersomnia.
Treatment of hypersomnia/narcolepsy should be individualized and pharmacologic intervention with stimulants may be beneficial.
In a baseline evaluation of 82 childhood CNS tumor survivors (median age, 13.8 years) participating in a randomized controlled trial of neurofeedback, 48% of survivors endorsed sleep problems and scored significantly worse than the norm on the Sleep Disturbance Scale for Children in the subscales for initiating and maintaining sleep, excessive somnolence, and total scale.[106]
Emotional problems and/or hyperactivity/inattention were independent potential risk factors for sleep problems. Sleep problems were also associated with worse parent-reported executive functioning.
Other neurological sequelae
In a report from the CCSS that compared self-reported neurological late effects among 4,151 adult survivors of childhood ALL with siblings, survivors were at elevated risk for late-onset coordination problems, motor problems, seizures, and headaches.[91]
The overall cumulative incidence was 44% at 20 years. Serious headaches were most common, with a cumulative incidence of 25.8% at 20 years, followed by focal neurological dysfunction (21.2%) and seizures (7%).
Children who were treated with regimens that included cranial irradiation for ALL and those who suffered relapse were at increased risk for late-onset neurological sequelae.
A cross-sectional study evaluated neurological morbidity and quality of life in 162 survivors of childhood ALL (median age at evaluation, 15.7 years; median time from completion of therapy, 7.4 years) in concert with a clinical neurological examination.[107]
Neurological symptoms were present in 83% of survivors, but symptom-related morbidity was low and quality of life was high in most survivors.
The most commonly reported symptoms included neuropathy (63%), headache (46.9%), dizziness (33.3%), and back pain (22.8%).
Female sex, ten doses or more of intrathecal chemotherapy, cranial irradiation, CNS leukemia at diagnosis, and history of ALL relapse were associated with neurological morbidity.
Neuroimaging studies of irradiated and nonirradiated ALL survivors demonstrate a variety of CNS abnormalities, including leukoencephalopathy, cerebral lacunes, cerebral atrophy, and dystrophic calcifications (mineralizing microangiopathy).[53,59,108,109]
Among these, abnormalities of cerebral white matter integrity and volume have been correlated with neurocognitive outcomes.
Cavernomas have also been observed in ALL survivors treated with cranial irradiation. They have been speculated to result from angiogenic processes as opposed to tumorigenesis.[110]
In a retrospective cross-sectional study, 101 survivors of childhood ALL (mean time since diagnosis, 27.6 years) who were treated with cranial irradiation (63.4% received ≤18 Gy) underwent neurocognitive testing and 3T brain MRI.[111]
Small focal intracerebral hemorrhages that are only visible on exquisitely sensitive MRI sequences were identified and localized using susceptibility weighted imaging.
At least one microbleed was present in 85% of survivors, and they occurred more frequently in frontal lobes.
A radiation dose of 24 Gy conveyed a fivefold greater risk of having multiple microbleeds when compared with a dose of 18 Gy.
No significant difference was found in neurocognitive scores with either the absence or presence of microbleeds or their location.
CCSS investigators evaluated treatment-related neurological sequelae in survivors of childhood CNS tumors.[90]
In 1,876 5-year survivors of CNS tumors from the CCSS, the cumulative incidence of headaches increased from 38% at 5 years to 53% at 30 years since diagnosis.
Coordination problems increased from 21% at 5 years to 53% at 30 years since diagnosis, and motor impairment increased from 21% to 35% during this same time period.
Increased risk of motor impairment was associated with tumor recurrence (HR, 2.6), development of a meningioma (HR, 2.3), and stroke (HR, 14.9).
The cumulative incidence of hearing loss increased from 9% at 5 years to 23% at 30 years, cumulative incidence of tinnitus increased from 8% at 5 years to 21% at 30 years, and cumulative incidence of vertigo increased from 9% at 5 years to 17% at 30 years.
Risks of motor impairment (HR, 7.6) and hearing loss (HR, 18.4) were elevated compared with siblings.
CCSS investigators estimated the prevalence and cumulative incidence of neuromuscular dysfunction (motor or sensory dysfunction) among 25,583 childhood cancer survivors and 5,044 siblings.[112]
Neuromuscular dysfunction was prevalent in 14.7% of survivors 5 years after diagnosis, compared with 1.5% in siblings (prevalence ratio [PR], 9.9%; 95% CI, 7.9–12.4).
The prevalence of neuromuscular dysfunction was highest in survivors of CNS tumors (PR, 27.6; 95% CI, 22.1–34.6) and sarcomas (PR, 11.5; 95% CI, 9.1–14.5).
The 20-year cumulative incidence increased to 24.3% in survivors 20 years after diagnosis.
Cancer treatments associated with increased prevalence of neuromuscular dysfunction included spinal radiation therapy, increasing doses of cranial radiation therapy, and platinum agents.
Neuromuscular dysfunction was associated with downstream adverse outcomes, including concurrent or later obesity (PR, 1.1; 95% CI, 1.1–1.2), anxiety (PR, 2.5; 95% CI, 2.2–2.9), depression (PR, 2.1; 95% CI, 1.9–2.3), and lower likelihood of graduating college (PR, 0.92; 95% CI, 0.90–0.94) and finding employment (PR, 0.8; 95% CI, 0.8–0.9).
SJLIFE investigators assessed motor and sensory impairment in 378 survivors of childhood CNS tumors (5 years or longer from diagnosis) and 445 age-, sex-, and race-matched controls, using the modified Total Neuropathy Score.[113]
Among survivors, 91 (24.1%; 95% CI, 19.8%–29.4%) had any grade 2 or higher motor or sensory impairment (motor only, 27 [7.1%]; sensory only, 40 [10.6%]; both, 24 [6.4%]).
Survivors were more likely than controls to have grade 2 or higher impairment (13.5% for survivors [7.7% for grade 2 and 5.8% for grade 3] vs. 0.9% for controls; P < .001).
Survivors of ependymoma had the highest prevalence of motor impairment (22.0%), and survivors of medulloblastoma had the highest prevalence of sensory impairment (33.3%).
Treatment factors associated with grade 2 or higher motor impairment in multivariable models included vinca alkaloid exposure of 15 mg/m2 or less compared with no exposure (OR, 4.38; 95% CI, 1.06–18.08) and etoposide exposure of more than 2,036 mg/m2 compared with no exposure (OR, 12.61; 95% CI, 2.19–72.72).
Radiation myelopathy is a rare but severe complication of radiation therapy exposure. The PENTEC spinal cord task force examined the factors that potentially contribute to radiation myelitis in the pediatric population.[114]
Thirty-two patients (treated between 1962 and 2002) with radiation myelopathy were identified, for a crude incidence of 0.5%. The median age was 13 years, and 52% of the patients were male. No apparent association was identified for age at the time of radiation therapy. The most common cancer diagnoses were rhabdomyosarcoma, medulloblastoma, and Hodgkin lymphoma.
The median radiation dose was 40 Gy, and the fraction size was 1.8 Gy. The median latency period from completion of radiation therapy to development of radiation myelopathy was 7 months (range, 1–29 months), with higher radiation therapy doses correlating with longer latency periods (P = .03).
The most common chemotherapies given to patients with radiation myelopathy were vincristine, intrathecal (IT) methotrexate, and IT cytarabine. Ten patients received vincristine, IT methotrexate, and IT cytarabine.
Chemotherapy appears to reduce spinal cord tolerance to radiation. The radiation therapy dose resulting in radiation myelopathy was lower in patients who received chemotherapy than in those who did not receive chemotherapy (39.6 ± 8.7 Gy vs. 49.7 ± 8.2 Gy; P = .04).
Eight of the 32 cases of radiation myelopathy occurred in patients treated on a single Intergroup Rhabdomyosarcoma Study Group (treated with intensive IT methotrexate and cytarabine chemotherapy).
Many of the reports were from an era before CT-based treatment planning. Also, the investigators could not find any signs that the developing spinal cord is more sensitive than the adult spinal cord.
Table 4 summarizes CNS late effects and the related health screenings.
Table 4. Central Nervous System Late Effectsa
Predisposing Therapy
Neurological Effects
Health Screening
IQ = intelligence quotient; IT = intrathecal; IV = intravenous.
Many childhood cancer survivors report reduced quality of life, impaired health status, or other adverse psychosocial outcomes, compared with siblings or noncancer population groups.[115,116] Vulnerable groups have been identified related to sociodemographic factors (e.g., female sex), specific cancer diagnoses (e.g., CNS tumor), cancer treatments (e.g., cranial radiation therapy), health behaviors (e.g., smoking), and type/cumulative burden of chronic health conditions. The diagnosis of childhood cancer may also affect psychosocial outcomes and the expected attainment of functional and social independence in adulthood. Several investigations have demonstrated that survivors of pediatric CNS tumors are particularly vulnerable.[49,117]
Evidence for adverse psychosocial adjustment after childhood cancer has been derived from sources, ranging from patient-reported or proxy-reported outcomes to data from population-based registries. The former may be limited by small sample size, cohort selection and participation bias, and variable methods and venues (clinical vs. distance-based survey) of assessments. The latter is often not well correlated with clinical and treatment characteristics that permit the identification of survivors at high risk of psychosocial deficits.
Achievement of social milestones
Survivors with neurocognitive deficits are particularly vulnerable to deficits in achievement of expected social outcomes during adulthood.
Adult survivors of childhood cancer participating in the CCSS who were treated with CNS-directed therapies did not achieve independence at rates comparable with survivors who did not receive CNS-directed therapies or their sibling counterparts. Associations between neurological late effects and attainment of independence were examined in adult survivors of cancer treated with (n = 7,881) and without CNS-directed therapies (n = 8,039) and a sibling cohort (n = 3,994). Compared with the other groups, survivors of childhood cancer treated with CNS-directed therapies showed three distinct patterns of functional independence: (1) moderately independent, never married, and nonindependent living (78.7%); (2) moderately independent, unable to drive (15.6%); (3) not independent (5.7%). No survivors treated with CNS-directed therapies were fully independent (a fourth identified class). In contrast, 50% of survivors who did not receive CNS treatment and 60% of siblings were classified as fully independent. Nonindependence was associated with emotional distress symptoms (i.e., depression, anxiety, somatization, and suicidal ideation).[118]
In a population-based study of adult survivors of CNS tumors diagnosed in childhood or adolescence, survivors had significantly poorer self-perception and self-esteem than did individuals in the general population. Female sex, persistent visible physical sequelae, specific tumor type, and treatment with cranial radiation therapy predicted poor self-perception outcomes.[119]
In a series of CNS malignancy survivors (n = 802) reported from the CCSS, adverse outcome on multiple indicators of successful adult adjustment (educational achievement, income, employment, and marital status) were most prevalent among survivors who reported neurocognitive dysfunction.[42]
Collectively, studies evaluating psychosocial outcomes among CNS tumor survivors indicate deficits in social competence that worsen over time.[120] This includes problems with peer rejection and isolation in childhood/adolescence, and the inability to develop friendships and romantic relationships as adults.
In an SJLIFE study of 224 survivors of CNS tumors (median current age, 26 years; median time from diagnosis, 18 years), neurocognitive impairment was significantly associated with lower educational attainment, unemployment, and dependent living.[3]
Among one subgroup, significant impairments in social perceptions skills, including affect recognition, contributed to limited achievement of functional outcomes. Self-reports of social adjustment were generally within normal limits, suggesting limited self-insight into social deficits.[121]
In a series of 1,560 adolescent survivors of childhood ALL treated with chemotherapy alone, the CCSS identified a significant proportion of survivors who still experienced problems with headstrong behavior, inattention-hyperactivity, and social withdrawal, which were associated with an increased risk of special education placement and predicted reduced adult educational attainment.[70]
In a cross-sectional study of 855 childhood leukemia survivors (mean, 10.2 years from diagnosis) in the Leucémie de l’Enfant et de l’Adolescent (LEA) cohort, investigators identified independent factors associated with repeating a grade in school, including low parental education and household financial difficulties, emphasizing the importance of considering socioeconomic factors that may affect access to educational support. Survivors who were adolescents (aged 11–17 years) at diagnosis were also at greater risk than were survivors who were children (aged <11 years) at diagnosis.[122]
In a population-based, cross-sectional, survivorship study, 28% of adolescent and young adult (AYA) survivors of sarcoma (aged 18–39 years at diagnosis; median time from diagnosis, 6.2 years) experienced impaired social functioning.[123] Multivariable analysis identified independent associations with impaired social functioning and unemployment (OR, 3.72; 95% CI, 1.26–10.97) and having to make lifestyle changes because of financial problems caused by an individual’s physical condition or medical treatment (OR, 3.39; 95% CI, 1.12–10.30). In contrast, better social support reduced the risk of impaired social functioning (OR, 0.74; 95% CI, 0.57–0.96).
Psychological distress and suicidality
Childhood cancer survivors are also at risk of developing symptoms of psychological distress and suicidality.[124]
A CCSS report characterized the prevalence of and risk factors for self-reported emotional distress and HRQOL in survivors of rhabdomyosarcoma (n = 713) and their siblings (n = 706). The study also identified risk factors associated with these outcomes in survivors.[78]
Compared with their siblings, more survivors reported elevated emotional distress (somatic distress: 12.9% vs. 4.7%; anxiety: 11.7% vs. 5.9%; depression: 22.8% vs. 16.9%) and poorer HRQOL (physical functioning: 11.1% vs. 2.8%; role functioning due to physical problems: 16.8% vs. 8.2%; pain: 17.5% vs. 10.0%; vitality: 22.3% vs. 13.8%; social functioning: 14.4% vs. 6.8%; emotional functioning: 17.1% vs. 10.6%).
Chest and pelvic radiation exposure predicted an increased risk of impaired physical functioning (OR, 2.68 and 3.44, respectively).
Smoking was associated with anxiety (OR, 2.71) and depression (OR, 1.77).
Neurological conditions increased the risk of anxiety (OR, 2.30), and hearing conditions increased the risk of depression (OR, 1.79).
The SJLIFE study was used to compare the risks of suicidal ideation, suicidal behaviors, and mortality in adult survivors of childhood cancer with those of the general population.[125]
Survivors (n = 3,096) reported a similar 12-month prevalence of suicidal ideation compared with the general population (standardized incidence ratio [SIR], 0.68) and a lower prevalence of suicidal behaviors (planning: SIR, 0.17; attempts: SIR, 0.07) and mortality (standardized mortality ratio, 0.60).
Among survivors, depression (RR, 12.30), anxiety (RR, 2.19), and financial stress (RR, 1.47) were found to be risk factors associated with suicidal ideation.
Survivors who were currently single, widowed, or divorced; who were not working full time; who reported financial stress during the previous year; and who reported sleep disturbances had an approximately 30% to 50% higher prevalence of suicidal ideation.
A CCSS study evaluated the prevalence of recurrent suicidal ideation among 9,128 adult long-term survivors of childhood cancer.[126]
Survivors were more likely to report late suicidal ideation (OR, 1.9; 95% CI, 1.5–2.5) and recurrent suicidal ideation (OR, 2.6; 95% CI, 1.8–3.8) compared with siblings.
History of seizure was associated with a twofold increased likelihood of suicide ideation in survivors.
A population-based study evaluated suicide among adults treated for cancer before age 25 years.[127]
The absolute risk of suicide was low (24 cases among 3,375 deaths).
The HR of suicide was increased among individuals treated for cancer in childhood (0–14 years; HR, 2.5; 95% CI, 1.7–3.8) and in adolescence and young adulthood (15–24 years; HR, 2.3; 95% CI, 1.2–4.6).
The presence of chronic health conditions can also impact aspects of psychological health.
In a study that evaluated psychological outcomes among long-term survivors treated with HSCT, 22% of survivors and 8% of sibling controls reported adverse outcomes. Somatic distress was the most prevalent condition and affected 15% of HSCT survivors, representing a threefold higher risk compared with siblings. HSCT survivors with severe or life-threatening health conditions and active chronic GVHD had a twofold increased risk of somatic distress.[128]
A report from the CCSS revealed that the presence of chronic pulmonary, endocrine, and cardiac conditions was associated with increased risk of psychological distress symptoms in a sample of 5,021 adult survivors of childhood cancer.[129]
In a CCSS investigation that evaluated long-term psychological and educational outcomes among survivors of neuroblastoma, survivors demonstrated elevated risks of psychological impairment, which was associated with the use of special education services and lower educational attainment. The presence of two or more chronic health conditions, but not common treatment exposures, predicted psychological impairment. Specifically, pulmonary disease predicted impairment in all five psychological domains, whereas endocrine disease and peripheral neuropathy each predicted impairment in three domains.[130]
Incorporation of psychological screening into clinical visits for childhood cancer survivors may be valuable. However, limiting such evaluations to those returning to long-term follow-up clinics may result in a biased subsample of survivors with more difficulties, and precise prevalence rates may be difficult to establish.
In a study of 101 adult survivors of childhood cancer, psychological screening was performed during a routine annual evaluation at the survivorship clinic at the Dana Farber Cancer Institute.[131]
On the Symptom Checklist 90 Revised, 32 survivors had a positive screen (indicating psychological distress), and 14 survivors reported at least one suicidal symptom.
Risk factors for psychological distress included survivors’ dissatisfaction with physical appearance, poor physical health, and treatment with cranial irradiation.
This instrument was shown to be feasible for use in the clinic-visit setting because the psychological screening was completed in less than 30 minutes and did not appear to cause distress in the survivors in 80% of cases.
A population-based study from Taiwan compared the prevalence of serious mental illnesses in 5,121 childhood and adolescent cancer survivors with that of population controls.[132]
Survivors (mean age, 9 years; 60% were 5 or more years from diagnosis) showed increased risks of six serious mental illnesses compared with controls. These disorders included the following:
Survivors were younger than controls at the time of diagnosis of ADHD, schizophrenia, major depressive disorder, and OCD.
Risks of serious mental illnesses varied according to specific cancer types. The greatest number of major psychiatric disorders was observed among survivors of brain cancer and lymphatic/hematopoietic tissue cancer.
A population-based study linked individuals with a history of six common cancers diagnosed at age 15 to 21 years to provincial health care data to compare rates of outpatient (family physician and psychiatrist) visits for psychiatric indications and time to severe psychiatric events (emergency room visit, hospitalization, and suicide). The study included 2,208 AYA cancer patients and 10,457 matched controls.[133]
Five-year AYA survivors experienced higher rates of outpatient mental health visits than did controls (671 vs. 506 visits per 1,000 person-years; RR, 1.3), higher risk of a severe psychiatric episode (HR, 1.2), and higher risk of a psychotic disorder–associated severe event (HR, 2.0).
Treatment in an adult center was associated with substantially higher outpatient visit rates compared with treatment in pediatric settings (RR, 1.8).
PTSD after childhood cancer
Despite the many stresses associated with the diagnosis of cancer and its treatment, studies have generally shown low levels of post-traumatic stress symptoms and PTSD in children with cancer, typically no higher than those in healthy comparison children.[134]
The prevalence of PTSD and post-traumatic stress symptoms has been reported in 15% to 20% of young adult survivors of childhood cancer, with estimates varying based on criteria used to define these conditions.[135]
In a cohort of 5,121 childhood and adolescent cancer survivors (mean age, 9 years), the incidence of PTSD was 0.59%. Compared with controls, survivors showed a 6.10-fold elevated risk of being diagnosed with PTSD. Brain cancer survivors had the highest risk of all cancer diagnoses for PTSD (HR, 18.50; 95% CI, 2.11–162.64).[132]
Patient and parent adaptive style appear to be significant determinants of PTSD in the pediatric oncology setting.[136,137]
Survivors with PTSD reported more psychological problems and negative beliefs about their illness and health status than did those without PTSD.[138,139]
A subset of adult survivors (9%) from the CCSS reported functional impairment and/or clinical distress in addition to the set of symptoms consistent with a full diagnosis of PTSD.[140]
PTSD was significantly more prevalent in survivors than in sibling comparisons.
PTSD was significantly associated with being unmarried, having an annual income of less than $20,000, being unemployed, having a high school education or less, and being older than 30 years.
Survivors who were treated with cranial irradiation before age 4 years were at particularly higher risk of developing PTSD.
Intensive cancer-directed therapy was also associated with increased risk of full PTSD.
Because avoidance of places and persons associated with the cancer is part of PTSD, the syndrome may interfere with obtaining appropriate health care.[141]
Those with PTSD perceive greater current threats to their lives or the lives of their children.
Other risk factors for PTSD include poor family functioning, decreased social support, and noncancer stressors.
Psychosocial outcomes among childhood, adolescent, and young adult cancer survivors
Most research on late effects after cancer has focused on individuals with a cancer diagnosis during childhood. Little is known about the specific impact of a cancer diagnosis with an onset in adolescence or the impact of childhood cancer on AYA psychosocial outcomes.
Evidence (psychosocial outcomes in AYA cancer survivors):
Adult survivors of cancer diagnosed during adolescence (aged 15–18 years) (N = 825) were compared with an age-matched sample from the general population and a comparison group of adults without cancer.[142]
Female survivors of adolescent cancers achieved fewer developmental milestones related to their psychosexual development, such as having their first boyfriend, or they reached these milestones later.
Male survivors were more likely to live with their parents than were same-sex controls.
Adolescent cancer survivors were less likely to have ever married or have had children. Survivors were significantly older at their first marriage and at the birth of their first child than were their age-matched samples.
Survivors in this cohort were also significantly less satisfied with their general and health-related life than were people in a community-based control group. Impaired general and health-related life satisfaction were associated with somatic late effects, symptoms of depression and anxiety, and lower rates of posttraumatic growth.[143]
A survey of 4,054 AYA cancer survivors and 345,592 respondents who had no history of cancer reported the following:[144]
AYA cancer survivors were more likely to smoke (26% vs. 18%), be affected by obesity (31% vs. 27%), and have chronic conditions such as cardiovascular disease (14% vs. 7%), hypertension (35% vs. 9%), asthma (15% vs. 8%), disability (36% vs. 18%), and poor mental health (20% vs. 10%).
They were also less likely to receive medical care because of cost (24% vs. 15%).
The CCSS evaluated outcomes of 2,979 adolescent survivors and 649 siblings of childhood cancer survivors to determine the incidence of difficulty in six behavioral and social domains (depression/anxiety, being headstrong, attention deficit, peer conflict/social withdrawal, antisocial behaviors, and social competence).[145]
Survivors were 1.5 times (95% CI, 1.1–2.1) more likely than siblings to have symptoms of depression/anxiety and 1.7 times (95% CI, 1.3–2.2) more likely than siblings to have antisocial behaviors.
Scores in the depression/anxiety, attention deficit, and antisocial domains were significantly elevated in adolescents treated for leukemia or CNS tumors, compared with the scores in siblings.
In addition, survivors of neuroblastoma had difficulty in the depression/anxiety and antisocial domains.
CNS-directed treatments (cranial radiation therapy and/or intrathecal methotrexate) were specific risk factors for adverse behavioral outcomes.
Another CCSS study evaluated psychological and neurocognitive function in 2,589 long-term cancer survivors who were diagnosed during adolescence and young adulthood.[146]
Compared with a sibling cohort, survivors diagnosed during adolescence and young adulthood reported higher rates of depression (OR, 1.55; 95% CI, 1.04–2.30) and anxiety (OR, 2.00; 95% CI, 1.17–3.43) and reported more cognitive problems affecting task efficiency (OR, 1.72; 95% CI, 1.21–2.43), emotional regulation (OR, 1.74; 95% CI, 1.26–2.40), and memory (OR, 1.44; 95% CI, 1.09–1.89).
Survivors of lymphoma and sarcoma diagnosed during later adolescence were at reduced risk of psychosocial and neurocognitive problems than were those diagnosed before age 11 years. These outcomes did not differ by age at diagnosis among CNS tumor and leukemia survivors.
Survivors diagnosed during adolescence and young adulthood were also significantly less likely than sibling controls to have attained a post–high school education, be working full time, be married, or be living independently; inferior social outcomes were related to neurocognitive symptoms.
A follow-up CCSS study evaluated profiles of symptom comorbidities in 3,993 adolescents (aged 13–17 years) treated for cancer.[147] Latent profile analysis identified four symptom profiles:
No significant symptoms.
Elevated internalizing symptoms (anxiety and/or depression, social withdrawal, and attention problems).
Elevated externalizing symptoms (headstrong behavior and attention problems).
Elevated internalizing and externalizing symptoms.
Overall results support that behavioral, emotional, and social symptoms frequently co-occur and are associated with treatment exposures (cranial radiation, corticosteroids, and methotrexate) and late effects (obesity, cancer-related pain, and sensory impairments) in adolescent survivors diagnosed between 1970 and 1986.
Another CCSS study characterized the prevalence and risk of pain, clinically significant interference in daily activities because of pain, and recurrent pain in 10,012 adult survivors of childhood cancer (median time since diagnosis, 23 years).[148]
A significant minority of survivors endorsed moderate to severe pain (29%), moderate to extreme pain interference (20%), and moderate to severe recurrent pain (9%).
Older age at diagnosis and follow-up, female sex, and presence of grades 3 to 4 chronic medical conditions were consistently associated with an increased risk of worse pain outcomes.
Minority racial and ethnic groups, diagnosis of CNS tumor, and treatment with platinum-based chemotherapy and cranial radiation were associated with an increased risk of late-occurrence pain and pain interference.
Depression and anxiety were associated with increased risk of all pain outcomes, and poor vitality mediated the effects of anxiety on high pain and pain interference.
An SJLIFE study evaluated the associations between comprehensive medical, neurocognitive, and physical performance assessments and self-reported pain, quality of life, and social functioning in 2,836 survivors (mean age, 32.2 years; mean time since diagnosis, 23.7 years) and 343 noncancer community controls.[149]
Moderate-to-very-severe pain with moderate-to-extreme daily interference was endorsed by 18% of survivors, compared with 8% of controls (P < .001).
Survivors of soft tissue sarcoma (OR, 9.25), non-Hodgkin lymphoma (OR, 4.13), and Ewing sarcoma and/or osteosarcoma (OR, 3.93) had the highest odds of pain with daily interference, compared with controls.
In multivariable models that included treatment exposures, histories of amputation (OR, 1.89) and/or limb-sparing surgery (OR, 2.30) were associated with increased odds of pain with daily interference.
Pain with daily interference was associated with an increased risk of impaired neurocognition (attention: RR, 1.88; memory: RR, 1.65) and physical functioning (aerobic capacity: RR, 2.29; mobility: RR, 1.71).
Pain with daily interference was also associated with an increased risk of impaired social functioning (inability to hold a job and/or attend school: RR, 4.46; assistance with routine and/or personal care needs: RR, 5.64), and health-related quality of life (physical: RR, 6.34; emotional: RR, 2.83).
A Nordic, register-based cohort study evaluated whether childhood cancer survivors (n = 18,621) were at a higher risk of developing psychiatric disorders later in life than their siblings (n = 24,775) and the general population (n = 88,630).[150]
The cumulative incidence of contact with a psychiatric hospital by age 30 years was 15.9% for childhood cancer survivors, 14.0% for siblings, and 12.7% for matched (by birth year, sex, and country/municipality) population controls.
The absolute difference was small, but survivors were still at a higher RR for any psychiatric hospital contact than their siblings (1.39) and matched controls (HR, 1.34).
A CCSS study evaluated associations between treatment changes and neurocognition as well as the impact of neurocognition and chronic health conditions on adult independence in 1,284 adult survivors of childhood gliomas (median age, 30 years at assessment and 22 years from diagnosis).[151]
Cranial radiation exposure decreased over the study period (1970s–1990s).
Survivors with any treatment exposures (surgery only, chemotherapy with or without surgery, cranial radiation therapy with or without chemotherapy/surgery) had an elevated risk of neurocognitive impairment, compared with siblings.
While most long-term glioma survivors in the cohort achieved adult independence, associations with treatment-related neurocognitive impairment and chronic health conditions were observed among those with functional nonindependence.
Evidence (functional and social independence):
In a study of 665 survivors of CNS tumors (54% male; 52% treated with cranial radiation therapy; median age, 15 years; and 12 years from diagnosis), CCSS investigators observed the following:[117]
Almost 50% of survivors experienced social difficulties related to peer relationships that exceeded those of survivors of solid tumors and sibling controls.
Cranial radiation exposure predicted impaired social and peer relationships, and cognitive impairment mediated these associations.
An SJLIFE study investigated functional and social independence in 306 CNS tumor survivors (astrocytoma [n = 130], medulloblastoma [n = 77], ependymoma [n = 36], and other [n = 63]; median age, 25 years; and time since diagnosis, 16.8 years).[49]
Only 40% of long-term survivors in the study cohort achieved complete independence as adults.
Predictors of nonindependence included treatment with craniospinal irradiation, history of hydrocephalus with shunting, and younger age at diagnosis.
Beyond impaired IQ scores, functional limitations in aerobic capacity, flexibility, and adaptive physical function were significantly associated with nonindependence.
References
Grill J, Kieffer V, Kalifa C: Measuring the neuro-cognitive side-effects of irradiation in children with brain tumors. Pediatr Blood Cancer 42 (5): 452-6, 2004. [PUBMED Abstract]
Marusak HA, Iadipaolo AS, Harper FW, et al.: Neurodevelopmental consequences of pediatric cancer and its treatment: applying an early adversity framework to understanding cognitive, behavioral, and emotional outcomes. Neuropsychol Rev 28 (2): 123-175, 2018. [PUBMED Abstract]
Brinkman TM, Krasin MJ, Liu W, et al.: Long-Term Neurocognitive Functioning and Social Attainment in Adult Survivors of Pediatric CNS Tumors: Results From the St Jude Lifetime Cohort Study. J Clin Oncol 34 (12): 1358-67, 2016. [PUBMED Abstract]
Moxon-Emre I, Bouffet E, Taylor MD, et al.: Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. J Clin Oncol 32 (17): 1760-8, 2014. [PUBMED Abstract]
Krull KR, Zhang N, Santucci A, et al.: Long-term decline in intelligence among adult survivors of childhood acute lymphoblastic leukemia treated with cranial radiation. Blood 122 (4): 550-3, 2013. [PUBMED Abstract]
Annett RD, Hile S, Bedrick E, et al.: Neuropsychological functioning of children treated for acute lymphoblastic leukemia: impact of whole brain radiation therapy. Psychooncology 24 (2): 181-9, 2015. [PUBMED Abstract]
van der Plas E, Qiu W, Nieman BJ, et al.: Sex-Specific Associations Between Chemotherapy, Chronic Conditions, and Neurocognitive Impairment in Acute Lymphoblastic Leukemia Survivors: A Report From the Childhood Cancer Survivor Study. J Natl Cancer Inst 113 (5): 588-596, 2021. [PUBMED Abstract]
Krull KR, Khan RB, Ness KK, et al.: Symptoms of attention-deficit/hyperactivity disorder in long-term survivors of childhood leukemia. Pediatr Blood Cancer 57 (7): 1191-6, 2011. [PUBMED Abstract]
Kahalley LS, Conklin HM, Tyc VL, et al.: ADHD and secondary ADHD criteria fail to identify many at-risk survivors of pediatric ALL and brain tumor. Pediatr Blood Cancer 57 (1): 110-8, 2011. [PUBMED Abstract]
Mahajan A, Stavinoha PL, Rongthong W, et al.: Neurocognitive Effects and Necrosis in Childhood Cancer Survivors Treated With Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 401-416, 2024. [PUBMED Abstract]
Cheung YT, Brinkman TM, Li C, et al.: Chronic Health Conditions and Neurocognitive Function in Aging Survivors of Childhood Cancer: A Report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 110 (4): 411-419, 2018. [PUBMED Abstract]
Bass JK, Liu W, Banerjee P, et al.: Association of Hearing Impairment With Neurocognition in Survivors of Childhood Cancer. JAMA Oncol 6 (9): 1363-1371, 2020. [PUBMED Abstract]
Barlow-Krelina E, Chen Y, Yasui Y, et al.: Consistent Physical Activity and Future Neurocognitive Problems in Adult Survivors of Childhood Cancers: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 38 (18): 2041-2052, 2020. [PUBMED Abstract]
Bernal JDK, Recchia F, Yu DJ, et al.: Physical activity and exercise for cancer-related cognitive impairment among individuals affected by childhood cancer: a systematic review and meta-analysis. Lancet Child Adolesc Health 7 (1): 47-58, 2023. [PUBMED Abstract]
Phillips NS, Stratton KL, Williams AM, et al.: Late-onset Cognitive Impairment and Modifiable Risk Factors in Adult Childhood Cancer Survivors. JAMA Netw Open 6 (5): e2316077, 2023. [PUBMED Abstract]
Traunwieser T, Loos E, Ottensmeier H, et al.: Survivors of infant atypical teratoid/rhabdoid tumors present with severely impaired cognitive functions especially for fluid intelligence and visual processing: data from the German brain tumor studies. Pediatr Blood Cancer 71 (5): e30910, 2024. [PUBMED Abstract]
Merchant TE, Schreiber JE, Wu S, et al.: Critical combinations of radiation dose and volume predict intelligence quotient and academic achievement scores after craniospinal irradiation in children with medulloblastoma. Int J Radiat Oncol Biol Phys 90 (3): 554-61, 2014. [PUBMED Abstract]
Armstrong GT, Conklin HM, Huang S, et al.: Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro Oncol 13 (2): 223-34, 2011. [PUBMED Abstract]
Di Pinto M, Conklin HM, Li C, et al.: Learning and memory following conformal radiation therapy for pediatric craniopharyngioma and low-grade glioma. Int J Radiat Oncol Biol Phys 84 (3): e363-9, 2012. [PUBMED Abstract]
Ris MD, Walsh K, Wallace D, et al.: Intellectual and academic outcome following two chemotherapy regimens and radiotherapy for average-risk medulloblastoma: COG A9961. Pediatr Blood Cancer 60 (8): 1350-7, 2013. [PUBMED Abstract]
Bowers DC, Liu Y, Leisenring W, et al.: Late-occurring stroke among long-term survivors of childhood leukemia and brain tumors: a report from the Childhood Cancer Survivor Study. J Clin Oncol 24 (33): 5277-82, 2006. [PUBMED Abstract]
Nassar SL, Conklin HM, Zhou Y, et al.: Neurocognitive outcomes among children who experienced seizures during treatment for acute lymphoblastic leukemia. Pediatr Blood Cancer 64 (8): , 2017. [PUBMED Abstract]
Torres VA, Ashford JM, Wright E, et al.: The impact of socioeconomic status (SES) on cognitive outcomes following radiotherapy for pediatric brain tumors: a prospective, longitudinal trial. Neuro Oncol 23 (7): 1173-1182, 2021. [PUBMED Abstract]
Mabbott DJ, Spiegler BJ, Greenberg ML, et al.: Serial evaluation of academic and behavioral outcome after treatment with cranial radiation in childhood. J Clin Oncol 23 (10): 2256-63, 2005. [PUBMED Abstract]
Brière ME, Scott JG, McNall-Knapp RY, et al.: Cognitive outcome in pediatric brain tumor survivors: delayed attention deficit at long-term follow-up. Pediatr Blood Cancer 50 (2): 337-40, 2008. [PUBMED Abstract]
Edelstein K, Spiegler BJ, Fung S, et al.: Early aging in adult survivors of childhood medulloblastoma: long-term neurocognitive, functional, and physical outcomes. Neuro Oncol 13 (5): 536-45, 2011. [PUBMED Abstract]
Palmer SL, Goloubeva O, Reddick WE, et al.: Patterns of intellectual development among survivors of pediatric medulloblastoma: a longitudinal analysis. J Clin Oncol 19 (8): 2302-8, 2001. [PUBMED Abstract]
Mulhern RK, Merchant TE, Gajjar A, et al.: Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol 5 (7): 399-408, 2004. [PUBMED Abstract]
Brinkman TM, Reddick WE, Luxton J, et al.: Cerebral white matter integrity and executive function in adult survivors of childhood medulloblastoma. Neuro Oncol 14 (Suppl 4): iv25-36, 2012. [PUBMED Abstract]
Jacola LM, Ashford JM, Reddick WE, et al.: The relationship between working memory and cerebral white matter volume in survivors of childhood brain tumors treated with conformal radiation therapy. J Neurooncol 119 (1): 197-205, 2014. [PUBMED Abstract]
Palmer SL, Glass JO, Li Y, et al.: White matter integrity is associated with cognitive processing in patients treated for a posterior fossa brain tumor. Neuro Oncol 14 (9): 1185-93, 2012. [PUBMED Abstract]
Ali JS, Ashford JM, Swain MA, et al.: Predictors of Cognitive Performance Among Infants Treated for Brain Tumors: Findings From a Multisite, Prospective, Longitudinal Trial. J Clin Oncol 39 (21): 2350-2358, 2021. [PUBMED Abstract]
Merchant TE, Conklin HM, Wu S, et al.: Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol 27 (22): 3691-7, 2009. [PUBMED Abstract]
Liu APY, Hastings C, Wu S, et al.: Treatment burden and long-term health deficits of patients with low-grade gliomas or glioneuronal tumors diagnosed during the first year of life. Cancer 125 (7): 1163-1175, 2019. [PUBMED Abstract]
Palmer SL, Armstrong C, Onar-Thomas A, et al.: Processing speed, attention, and working memory after treatment for medulloblastoma: an international, prospective, and longitudinal study. J Clin Oncol 31 (28): 3494-500, 2013. [PUBMED Abstract]
Knight SJ, Conklin HM, Palmer SL, et al.: Working memory abilities among children treated for medulloblastoma: parent report and child performance. J Pediatr Psychol 39 (5): 501-11, 2014. [PUBMED Abstract]
Schreiber JE, Gurney JG, Palmer SL, et al.: Examination of risk factors for intellectual and academic outcomes following treatment for pediatric medulloblastoma. Neuro Oncol 16 (8): 1129-36, 2014. [PUBMED Abstract]
Khan RB, Patay Z, Klimo P, et al.: Clinical features, neurologic recovery, and risk factors of postoperative posterior fossa syndrome and delayed recovery: a prospective study. Neuro Oncol 23 (9): 1586-1596, 2021. [PUBMED Abstract]
Schreiber JE, Palmer SL, Conklin HM, et al.: Posterior fossa syndrome and long-term neuropsychological outcomes among children treated for medulloblastoma on a multi-institutional, prospective study. Neuro Oncol 19 (12): 1673-1682, 2017. [PUBMED Abstract]
Moxon-Emre I, Taylor MD, Bouffet E, et al.: Intellectual Outcome in Molecular Subgroups of Medulloblastoma. J Clin Oncol 34 (34): 4161-4170, 2016. [PUBMED Abstract]
Acharya S, Wu S, Ashford JM, et al.: Association between hippocampal dose and memory in survivors of childhood or adolescent low-grade glioma: a 10-year neurocognitive longitudinal study. Neuro Oncol 21 (9): 1175-1183, 2019. [PUBMED Abstract]
Ellenberg L, Liu Q, Gioia G, et al.: Neurocognitive status in long-term survivors of childhood CNS malignancies: a report from the Childhood Cancer Survivor Study. Neuropsychology 23 (6): 705-17, 2009. [PUBMED Abstract]
Armstrong GT, Liu Q, Yasui Y, et al.: Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. J Natl Cancer Inst 101 (13): 946-58, 2009. [PUBMED Abstract]
Ris MD, Leisenring WM, Goodman P, et al.: Neuropsychological and socioeconomic outcomes in adult survivors of pediatric low-grade glioma. Cancer 125 (17): 3050-3058, 2019. [PUBMED Abstract]
Shabason EK, Brodsky C, Baran J, et al.: Clinical diagnosis of attention-deficit/hyperactivity disorder in survivors of pediatric brain tumors. J Neurooncol 143 (2): 305-312, 2019. [PUBMED Abstract]
Brinkman TM, Palmer SL, Chen S, et al.: Parent-reported social outcomes after treatment for pediatric embryonal tumors: a prospective longitudinal study. J Clin Oncol 30 (33): 4134-40, 2012. [PUBMED Abstract]
Moyer KH, Willard VW, Gross AM, et al.: The impact of attention on social functioning in survivors of pediatric acute lymphoblastic leukemia and brain tumors. Pediatr Blood Cancer 59 (7): 1290-5, 2012. [PUBMED Abstract]
Mitby PA, Robison LL, Whitton JA, et al.: Utilization of special education services and educational attainment among long-term survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. Cancer 97 (4): 1115-26, 2003. [PUBMED Abstract]
Brinkman TM, Ness KK, Li Z, et al.: Attainment of Functional and Social Independence in Adult Survivors of Pediatric CNS Tumors: A Report From the St Jude Lifetime Cohort Study. J Clin Oncol 36 (27): 2762-2769, 2018. [PUBMED Abstract]
Kahalley LS, Ris MD, Grosshans DR, et al.: Comparing Intelligence Quotient Change After Treatment With Proton Versus Photon Radiation Therapy for Pediatric Brain Tumors. J Clin Oncol 34 (10): 1043-9, 2016. [PUBMED Abstract]
Pulsifer MB, Sethi RV, Kuhlthau KA, et al.: Early Cognitive Outcomes Following Proton Radiation in Pediatric Patients With Brain and Central Nervous System Tumors. Int J Radiat Oncol Biol Phys 93 (2): 400-7, 2015. [PUBMED Abstract]
Kahalley LS, Peterson R, Ris MD, et al.: Superior Intellectual Outcomes After Proton Radiotherapy Compared With Photon Radiotherapy for Pediatric Medulloblastoma. J Clin Oncol 38 (5): 454-461, 2020. [PUBMED Abstract]
Reddick WE, Glass JO, Helton KJ, et al.: Prevalence of leukoencephalopathy in children treated for acute lymphoblastic leukemia with high-dose methotrexate. AJNR Am J Neuroradiol 26 (5): 1263-9, 2005. [PUBMED Abstract]
Waber DP, Queally JT, Catania L, et al.: Neuropsychological outcomes of standard risk and high risk patients treated for acute lymphoblastic leukemia on Dana-Farber ALL consortium protocol 95-01 at 5 years post-diagnosis. Pediatr Blood Cancer 58 (5): 758-65, 2012. [PUBMED Abstract]
Krull KR, Brinkman TM, Li C, et al.: Neurocognitive outcomes decades after treatment for childhood acute lymphoblastic leukemia: a report from the St Jude lifetime cohort study. J Clin Oncol 31 (35): 4407-15, 2013. [PUBMED Abstract]
Reddick WE, Shan ZY, Glass JO, et al.: Smaller white-matter volumes are associated with larger deficits in attention and learning among long-term survivors of acute lymphoblastic leukemia. Cancer 106 (4): 941-9, 2006. [PUBMED Abstract]
Kadan-Lottick NS, Zeltzer LK, Liu Q, et al.: Neurocognitive functioning in adult survivors of childhood non-central nervous system cancers. J Natl Cancer Inst 102 (12): 881-93, 2010. [PUBMED Abstract]
Schuitema I, Deprez S, Van Hecke W, et al.: Accelerated aging, decreased white matter integrity, and associated neuropsychological dysfunction 25 years after pediatric lymphoid malignancies. J Clin Oncol 31 (27): 3378-88, 2013. [PUBMED Abstract]
Armstrong GT, Reddick WE, Petersen RC, et al.: Evaluation of memory impairment in aging adult survivors of childhood acute lymphoblastic leukemia treated with cranial radiotherapy. J Natl Cancer Inst 105 (12): 899-907, 2013. [PUBMED Abstract]
Spiegler BJ, Kennedy K, Maze R, et al.: Comparison of long-term neurocognitive outcomes in young children with acute lymphoblastic leukemia treated with cranial radiation or high-dose or very high-dose intravenous methotrexate. J Clin Oncol 24 (24): 3858-64, 2006. [PUBMED Abstract]
Campbell LK, Scaduto M, Sharp W, et al.: A meta-analysis of the neurocognitive sequelae of treatment for childhood acute lymphocytic leukemia. Pediatr Blood Cancer 49 (1): 65-73, 2007. [PUBMED Abstract]
Cheung YT, Sabin ND, Reddick WE, et al.: Leukoencephalopathy and long-term neurobehavioural, neurocognitive, and brain imaging outcomes in survivors of childhood acute lymphoblastic leukaemia treated with chemotherapy: a longitudinal analysis. Lancet Haematol 3 (10): e456-e466, 2016. [PUBMED Abstract]
Jansen NC, Kingma A, Schuitema A, et al.: Neuropsychological outcome in chemotherapy-only-treated children with acute lymphoblastic leukemia. J Clin Oncol 26 (18): 3025-30, 2008. [PUBMED Abstract]
Iyer NS, Balsamo LM, Bracken MB, et al.: Chemotherapy-only treatment effects on long-term neurocognitive functioning in childhood ALL survivors: a review and meta-analysis. Blood 126 (3): 346-53, 2015. [PUBMED Abstract]
Jacola LM, Krull KR, Pui CH, et al.: Longitudinal Assessment of Neurocognitive Outcomes in Survivors of Childhood Acute Lymphoblastic Leukemia Treated on a Contemporary Chemotherapy Protocol. J Clin Oncol 34 (11): 1239-47, 2016. [PUBMED Abstract]
Espy KA, Moore IM, Kaufmann PM, et al.: Chemotherapeutic CNS prophylaxis and neuropsychologic change in children with acute lymphoblastic leukemia: a prospective study. J Pediatr Psychol 26 (1): 1-9, 2001 Jan-Feb. [PUBMED Abstract]
Buizer AI, de Sonneville LM, Veerman AJ: Effects of chemotherapy on neurocognitive function in children with acute lymphoblastic leukemia: a critical review of the literature. Pediatr Blood Cancer 52 (4): 447-54, 2009. [PUBMED Abstract]
Gandy K, Sapkota Y, Scoggins MA, et al.: Genetic variants, neurocognitive outcomes, and functional neuroimaging in survivors of childhood acute lymphoblastic leukemia. JNCI Cancer Spectr 7 (4): , 2023. [PUBMED Abstract]
Cole PD, Finkelstein Y, Stevenson KE, et al.: Polymorphisms in Genes Related to Oxidative Stress Are Associated With Inferior Cognitive Function After Therapy for Childhood Acute Lymphoblastic Leukemia. J Clin Oncol 33 (19): 2205-11, 2015. [PUBMED Abstract]
Jacola LM, Edelstein K, Liu W, et al.: Cognitive, behaviour, and academic functioning in adolescent and young adult survivors of childhood acute lymphoblastic leukaemia: a report from the Childhood Cancer Survivor Study. Lancet Psychiatry 3 (10): 965-972, 2016. [PUBMED Abstract]
Jacola LM, Conklin HM, Krull KR, et al.: The Impact of Intensified CNS-Directed Therapy on Neurocognitive Outcomes in Survivors of Childhood Acute Lymphoblastic Leukemia Treated Without Cranial Irradiation. J Clin Oncol 40 (36): 4218-4227, 2022. [PUBMED Abstract]
Krull KR, Cheung YT, Liu W, et al.: Chemotherapy Pharmacodynamics and Neuroimaging and Neurocognitive Outcomes in Long-Term Survivors of Childhood Acute Lymphoblastic Leukemia. J Clin Oncol 34 (22): 2644-53, 2016. [PUBMED Abstract]
Halsey C, Buck G, Richards S, et al.: The impact of therapy for childhood acute lymphoblastic leukaemia on intelligence quotients; results of the risk-stratified randomized central nervous system treatment trial MRC UKALL XI. J Hematol Oncol 4: 42, 2011. [PUBMED Abstract]
Kadan-Lottick NS, Brouwers P, Breiger D, et al.: A comparison of neurocognitive functioning in children previously randomized to dexamethasone or prednisone in the treatment of childhood acute lymphoblastic leukemia. Blood 114 (9): 1746-52, 2009. [PUBMED Abstract]
Williams AM, Cheung YT, Hyun G, et al.: Childhood Neurotoxicity and Brain Resilience to Adverse Events during Adulthood. Ann Neurol 89 (3): 534-545, 2021. [PUBMED Abstract]
Stefanski KJ, Anixt JS, Goodman P, et al.: Long-Term Neurocognitive and Psychosocial Outcomes After Acute Myeloid Leukemia: A Childhood Cancer Survivor Study Report. J Natl Cancer Inst 113 (4): 481-495, 2021. [PUBMED Abstract]
Edelmann MN, Daryani VM, Bishop MW, et al.: Neurocognitive and Patient-Reported Outcomes in Adult Survivors of Childhood Osteosarcoma. JAMA Oncol 2 (2): 201-8, 2016. [PUBMED Abstract]
van der Plas E, Darji H, Srivastava DK, et al.: Risk factors for neurocognitive impairment, emotional distress, and poor quality of life in survivors of pediatric rhabdomyosarcoma: A report from the Childhood Cancer Survivor Study. Cancer 130 (12): 2224-2236, 2024. [PUBMED Abstract]
Tonning Olsson I, Brinkman TM, Wang M, et al.: Neurocognitive and psychosocial outcomes in adult survivors of childhood soft-tissue sarcoma: A report from the St. Jude Lifetime Cohort. Cancer 126 (7): 1576-1584, 2020. [PUBMED Abstract]
Tonning Olsson I, Brinkman TM, Hyun G, et al.: Neurocognitive outcomes in long-term survivors of Wilms tumor: a report from the St. Jude Lifetime Cohort. J Cancer Surviv 13 (4): 570-579, 2019. [PUBMED Abstract]
Willard VW, Qaddoumi I, Chen S, et al.: Developmental and adaptive functioning in children with retinoblastoma: a longitudinal investigation. J Clin Oncol 32 (25): 2788-93, 2014. [PUBMED Abstract]
Willard VW, Qaddoumi I, Pan H, et al.: Cognitive and Adaptive Functioning in Youth With Retinoblastoma: A Longitudinal Investigation Through 10 Years of Age. J Clin Oncol 39 (24): 2676-2684, 2021. [PUBMED Abstract]
Brinkman TM, Merchant TE, Li Z, et al.: Cognitive function and social attainment in adult survivors of retinoblastoma: a report from the St. Jude Lifetime Cohort Study. Cancer 121 (1): 123-31, 2015. [PUBMED Abstract]
Ehrhardt MJ, Sandlund JT, Zhang N, et al.: Late outcomes of adult survivors of childhood non-Hodgkin lymphoma: A report from the St. Jude Lifetime Cohort Study. Pediatr Blood Cancer 64 (6): , 2017. [PUBMED Abstract]
Krull KR, Sabin ND, Reddick WE, et al.: Neurocognitive function and CNS integrity in adult survivors of childhood hodgkin lymphoma. J Clin Oncol 30 (29): 3618-24, 2012. [PUBMED Abstract]
Hesko C, Liu W, Srivastava DK, et al.: Neurocognitive outcomes in adult survivors of neuroblastoma: A report from the Childhood Cancer Survivor Study. Cancer 129 (18): 2904-2914, 2023. [PUBMED Abstract]
Wu NL, Phipps AI, Krull KR, et al.: Long-term patient-reported neurocognitive outcomes in adult survivors of hematopoietic cell transplant. Blood Adv 6 (14): 4347-4356, 2022. [PUBMED Abstract]
Willard VW, Leung W, Huang Q, et al.: Cognitive outcome after pediatric stem-cell transplantation: impact of age and total-body irradiation. J Clin Oncol 32 (35): 3982-8, 2014. [PUBMED Abstract]
Kenborg L, Winther JF, Linnet KM, et al.: Neurologic disorders in 4858 survivors of central nervous system tumors in childhood-an Adult Life after Childhood Cancer in Scandinavia (ALiCCS) study. Neuro Oncol 21 (1): 125-136, 2019. [PUBMED Abstract]
Wells EM, Ullrich NJ, Seidel K, et al.: Longitudinal assessment of late-onset neurologic conditions in survivors of childhood central nervous system tumors: a Childhood Cancer Survivor Study report. Neuro Oncol 20 (1): 132-142, 2018. [PUBMED Abstract]
Goldsby RE, Liu Q, Nathan PC, et al.: Late-occurring neurologic sequelae in adult survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. J Clin Oncol 28 (2): 324-31, 2010. [PUBMED Abstract]
Phillips NS, Khan RB, Li C, et al.: Seizures’ impact on cognition and quality of life in childhood cancer survivors. Cancer 128 (1): 180-191, 2022. [PUBMED Abstract]
Jain P, Gulati S, Seth R, et al.: Vincristine-induced neuropathy in childhood ALL (acute lymphoblastic leukemia) survivors: prevalence and electrophysiological characteristics. J Child Neurol 29 (7): 932-7, 2014. [PUBMED Abstract]
Ness KK, Jones KE, Smith WA, et al.: Chemotherapy-related neuropathic symptoms and functional impairment in adult survivors of extracranial solid tumors of childhood: results from the St. Jude Lifetime Cohort Study. Arch Phys Med Rehabil 94 (8): 1451-7, 2013. [PUBMED Abstract]
Kandula T, Farrar MA, Cohn RJ, et al.: Chemotherapy-Induced Peripheral Neuropathy in Long-term Survivors of Childhood Cancer: Clinical, Neurophysiological, Functional, and Patient-Reported Outcomes. JAMA Neurol 75 (8): 980-988, 2018. [PUBMED Abstract]
Rodwin RL, Ross WL, Rotatori J, et al.: Newly identified chemotherapy-induced peripheral neuropathy in a childhood cancer survivorship clinic. Pediatr Blood Cancer 69 (3): e29550, 2022. [PUBMED Abstract]
Varedi M, Lu L, Howell CR, et al.: Peripheral Neuropathy, Sensory Processing, and Balance in Survivors of Acute Lymphoblastic Leukemia. J Clin Oncol 36 (22): 2315-2322, 2018. [PUBMED Abstract]
Gurney JG, Kadan-Lottick NS, Packer RJ, et al.: Endocrine and cardiovascular late effects among adult survivors of childhood brain tumors: Childhood Cancer Survivor Study. Cancer 97 (3): 663-73, 2003. [PUBMED Abstract]
Ullrich NJ, Robertson R, Kinnamon DD, et al.: Moyamoya following cranial irradiation for primary brain tumors in children. Neurology 68 (12): 932-8, 2007. [PUBMED Abstract]
Wang C, Roberts KB, Bindra RS, et al.: Delayed cerebral vasculopathy following cranial radiation therapy for pediatric tumors. Pediatr Neurol 50 (6): 549-56, 2014. [PUBMED Abstract]
Mueller S, Fullerton HJ, Stratton K, et al.: Radiation, atherosclerotic risk factors, and stroke risk in survivors of pediatric cancer: a report from the Childhood Cancer Survivor Study. Int J Radiat Oncol Biol Phys 86 (4): 649-55, 2013. [PUBMED Abstract]
Reulen RC, Guha J, Bright CJ, et al.: Risk of cerebrovascular disease among 13 457 five-year survivors of childhood cancer: A population-based cohort study. Int J Cancer 148 (3): 572-583, 2021. [PUBMED Abstract]
Fullerton HJ, Stratton K, Mueller S, et al.: Recurrent stroke in childhood cancer survivors. Neurology 85 (12): 1056-64, 2015. [PUBMED Abstract]
Waxer JF, Wong K, Modiri A, et al.: Risk of Cerebrovascular Events Among Childhood and Adolescent Patients Receiving Cranial Radiation Therapy: A PENTEC Normal Tissue Outcomes Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 417-430, 2024. [PUBMED Abstract]
Khan RB, Merchant TE, Sadighi ZS, et al.: Prevalence, risk factors, and response to treatment for hypersomnia of central origin in survivors of childhood brain tumors. J Neurooncol 136 (2): 379-384, 2018. [PUBMED Abstract]
van Kooten JAMC, Maurice-Stam H, Schouten AYN, et al.: High occurrence of sleep problems in survivors of a childhood brain tumor with neurocognitive complaints: The association with psychosocial and behavioral executive functioning. Pediatr Blood Cancer 66 (11): e27947, 2019. [PUBMED Abstract]
Khan RB, Hudson MM, Ledet DS, et al.: Neurologic morbidity and quality of life in survivors of childhood acute lymphoblastic leukemia: a prospective cross-sectional study. J Cancer Surviv 8 (4): 688-96, 2014. [PUBMED Abstract]
Khong PL, Leung LH, Fung AS, et al.: White matter anisotropy in post-treatment childhood cancer survivors: preliminary evidence of association with neurocognitive function. J Clin Oncol 24 (6): 884-90, 2006. [PUBMED Abstract]
Zeller B, Tamnes CK, Kanellopoulos A, et al.: Reduced neuroanatomic volumes in long-term survivors of childhood acute lymphoblastic leukemia. J Clin Oncol 31 (17): 2078-85, 2013. [PUBMED Abstract]
Faraci M, Morana G, Bagnasco F, et al.: Magnetic resonance imaging in childhood leukemia survivors treated with cranial radiotherapy: a cross sectional, single center study. Pediatr Blood Cancer 57 (2): 240-6, 2011. [PUBMED Abstract]
Phillips NS, Hillenbrand CM, Mitrea BG, et al.: Cerebral microbleeds in adult survivors of childhood acute lymphoblastic leukemia treated with cranial radiation. Sci Rep 10 (1): 692, 2020. [PUBMED Abstract]
Rodwin RL, Chen Y, Yasui Y, et al.: Longitudinal Evaluation of Neuromuscular Dysfunction in Long-term Survivors of Childhood Cancer: A Report from the Childhood Cancer Survivor Study. Cancer Epidemiol Biomarkers Prev 30 (8): 1536-1545, 2021. [PUBMED Abstract]
Rodwin RL, Wang F, Lu L, et al.: Motor and sensory impairment in survivors of childhood central nervous system (CNS) tumors in the St. Jude Lifetime Cohort (SJLIFE). Cancer Med 13 (14): e7422, 2024. [PUBMED Abstract]
Cooper BT, Mayo CS, Milano MT, et al.: Predictive Factors Associated With Radiation Myelopathy in Pediatric Patients With Cancer: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 494-506, 2024. [PUBMED Abstract]
Ness KK, Hudson MM, Jones KE, et al.: Effect of Temporal Changes in Therapeutic Exposure on Self-reported Health Status in Childhood Cancer Survivors. Ann Intern Med 166 (2): 89-98, 2017. [PUBMED Abstract]
Ernst M, Hinz A, Brähler E, et al.: Quality of life after pediatric cancer: comparison of long-term childhood cancer survivors’ quality of life with a representative general population sample and associations with physical health and risk indicators. Health Qual Life Outcomes 21 (1): 65, 2023. [PUBMED Abstract]
Schulte F, Brinkman TM, Li C, et al.: Social adjustment in adolescent survivors of pediatric central nervous system tumors: A report from the Childhood Cancer Survivor Study. Cancer 124 (17): 3596-3608, 2018. [PUBMED Abstract]
Vuotto SC, Wang M, Okcu MF, et al.: Neurologic morbidity and functional independence in adult survivors of childhood cancer. Ann Clin Transl Neurol 11 (2): 291-301, 2024. [PUBMED Abstract]
Hörnquist L, Rickardsson J, Lannering B, et al.: Altered self-perception in adult survivors treated for a CNS tumor in childhood or adolescence: population-based outcomes compared with the general population. Neuro Oncol 17 (5): 733-40, 2015. [PUBMED Abstract]
Schulte F, Barrera M: Social competence in childhood brain tumor survivors: a comprehensive review. Support Care Cancer 18 (12): 1499-513, 2010. [PUBMED Abstract]
Papini C, Willard VW, Gajjar A, et al.: Social cognition and adjustment in adult survivors of pediatric central nervous system tumors. Cancer 129 (19): 3064-3075, 2023. [PUBMED Abstract]
Bonneau J, Berbis J, Michel G, et al.: Adolescence and Socioeconomic Factors: Key Factors in the Long-Term Impact of Leukemia on Scholastic Performance-A LEA Study. J Pediatr 205: 168-175.e2, 2019. [PUBMED Abstract]
Drabbe C, Coenraadts ES, van Houdt WJ, et al.: Impaired social functioning in adolescent and young adult sarcoma survivors: Prevalence and risk factors. Cancer 129 (9): 1419-1431, 2023. [PUBMED Abstract]
Ernst M, Brähler E, Wild PS, et al.: Risk factors for suicidal ideation in a large, registry-based sample of adult long-term childhood cancer survivors. J Affect Disord 265: 351-356, 2020. [PUBMED Abstract]
Lubas MM, Mirzaei Salehabadi S, Lavecchia J, et al.: Suicidality among adult survivors of childhood cancer: A report from the St. Jude Lifetime Cohort Study. Cancer 126 (24): 5347-5355, 2020. [PUBMED Abstract]
Brinkman TM, Zhang N, Recklitis CJ, et al.: Suicide ideation and associated mortality in adult survivors of childhood cancer. Cancer 120 (2): 271-7, 2014. [PUBMED Abstract]
Gunnes MW, Lie RT, Bjørge T, et al.: Suicide and violent deaths in survivors of cancer in childhood, adolescence and young adulthood-A national cohort study. Int J Cancer 140 (3): 575-580, 2017. [PUBMED Abstract]
Sun CL, Francisco L, Baker KS, et al.: Adverse psychological outcomes in long-term survivors of hematopoietic cell transplantation: a report from the Bone Marrow Transplant Survivor Study (BMTSS). Blood 118 (17): 4723-31, 2011. [PUBMED Abstract]
Vuotto SC, Krull KR, Li C, et al.: Impact of chronic disease on emotional distress in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 123 (3): 521-528, 2017. [PUBMED Abstract]
Zheng DJ, Krull KR, Chen Y, et al.: Long-term psychological and educational outcomes for survivors of neuroblastoma: A report from the Childhood Cancer Survivor Study. Cancer 124 (15): 3220-3230, 2018. [PUBMED Abstract]
Recklitis C, O’Leary T, Diller L: Utility of routine psychological screening in the childhood cancer survivor clinic. J Clin Oncol 21 (5): 787-92, 2003. [PUBMED Abstract]
Hsu TW, Liang CS, Tsai SJ, et al.: Risk of Major Psychiatric Disorders Among Children and Adolescents Surviving Malignancies: A Nationwide Longitudinal Study. J Clin Oncol 41 (11): 2054-2066, 2023. [PUBMED Abstract]
De R, Sutradhar R, Kurdyak P, et al.: Incidence and Predictors of Mental Health Outcomes Among Survivors of Adolescent and Young Adult Cancer: A Population-Based Study Using the IMPACT Cohort. J Clin Oncol 39 (9): 1010-1019, 2021.
Although cancer in children and adolescents is rare, it is the leading cause of death by disease after infancy among children in the United States (1). It is estimated that, in 2024, a total of 14,910 children and adolescents ages 0 to 19 will be diagnosed with cancer and 1,590 will die of the disease in the United States (1). Among children (ages 0 to 14 years), it is estimated that 9,620 will be diagnosed with cancer and 1040 will die of the disease (1). And among adolescents (ages 15 to 19 years), it is estimated that 5,290 will be diagnosed with cancer and 550 will die of the disease (1).
In the United States, the most common types of cancer diagnosed in 2016–2020 among children and adolescents were leukemias, malignant brain and other central nervous system (CNS) tumors, lymphomas, epithelial neoplasms and melanomas, soft tissue tumors, malignant germ cell tumors, and bone tumors.
The most common types of cancer among children and adolescents differed by age group. For example, leukemias were more common in those ages 1 to 4 than in other age groups and lymphomas were more common in those ages 15 to 19 than in other age groups.
Rates of the most common types of childhood and adolescent cancer also differ by race/ethnicity. For example, in 2017–2021, the incidence rate of leukemia was about twice as high in Hispanic and American Indian/Alaska Native children and adolescents as in Black children and adolescents. During the same time period, the rates of brain and other nervous system tumors were higher in White and American Indian/Alaska Native children and adolescents than in those of all other racial and ethnic groups.
What is the prognosis for children and adolescents with cancer?
The overall outlook for children and adolescents with cancer has improved greatly over the last half-century. In the mid-1970s, 58% of children (ages 0 to 14 years) and 68% of adolescents (ages 15 to 19 years) diagnosed with cancer survived at least 5 years (2). In 2013–2019, 5-year survival was 83.2% for children younger than 1 year, 87.8% for children ages 1–4 years, 85.7% for children ages 5–9 years, 85.5% for children ages 10–14 years, and 87.3% for adolescents ages 15–19 years.
Although the 5-year survival rates for most childhood cancers have improved in recent decades, the improvement has been especially dramatic for a few cancers, particularly acute lymphoblastic leukemia (ALL), the most common childhood cancer. Improved treatments for ALL introduced beginning in the 1960s and 1970s raised the 5-year survival rate for children diagnosed with this disease from 57% in 1975 to 92.3% in 2014–2020. The 5-year survival rate for children diagnosed with non-Hodgkin lymphoma also increased dramatically, from 43% in 1975 to 91.4% in 2014–2020.
In contrast, survival rates remain poor for some cancer types, for some age groups, and for some cancers within a site. For example:
Half of children with diffuse intrinsic pontine glioma (a rare type of brain tumor) survive less than 1 year from diagnosis, and only 10% survive 2 years from diagnosis (3).
In 2013–2019, the 5-year survival rate for lymphoid leukemias (mostly acute lymphoblastic leukemia) was 96%, 94%, and 86% for children ages 1–4, 5–9, and 10–14, respectively, compared with 61% for children younger than 1 year and 78% for adolescents.
Adolescents and young adults with ALL may have better outcomes if they are treated with pediatric treatment regimens than if they receive adult treatment regimens (4, 5). Possibly as a result of the increased use of these regimens, survival of adolescents and young adults with ALL has improved in recent years, even though their survival is still poorer than that of younger children with the disease (6).
The cancer mortality rate—the number of deaths due to cancer per 100,000 people per year—among children and adolescents younger than 20 years declined by more than 50% from 1975 to 2022. It dropped from 5.1 per 100,000 children and adolescents in 1975 to 2.2 per 100,000 children and adolescents in 2022.
However, despite the overall decrease in mortality, approximately 1,600 children and adolescents still die of cancer each year in the United States, indicating that new advances and continued research to identify effective treatments are required to further reduce childhood cancer mortality.
What are the possible causes of cancer in children and adolescents?
Most cancers in children, like those in adults, have alterations (changes, mutations, or variants) in genes that lead to uncontrolled cell growth and eventually cancer. Some genetic changes that are passed from parents to their children—known as germline variants—are associated with an increased risk of cancer. Genetic changes that lead to cancer can also occur spontaneously in cells during development.
Rare genetic changes that arise during development of one of the germ cells (sperm or egg) that unite to form the zygote that becomes a child can increase the risk of cancer in that child. Genetic changes can include broken, missing, rearranged, or extra chromosomes as well as gene variants. One such alteration is trisomy 21, or the presence of an extra copy of chromosome 21, which causes Down syndrome. Children with Down syndrome are 10–20 times more likely to develop leukemia than children without Down syndrome (11). However, only a very small proportion of childhood leukemia cases are linked to Down syndrome.
Environmental exposures
Genetic changes that may cause cancer can also occur in different cells of the body after birth, as the body is actively growing and developing during early childhood (12). The extent to which these changes reflect environmental exposures is unclear. Certain substances in the environment, such as cigarette smoke, asbestos, and ultraviolet (UV) radiation from the sun, are known to cause genetic changes that can lead to cancer. However, environmental causes of childhood cancer have been particularly difficult to identify, partly because cancer in children is rare and partly because it is difficult to determine what children might have been exposed to early in their development.
Nevertheless, several environmental exposures have been linked to childhood or adolescent cancer. For example, one study found that melanoma in children and adolescents (ages 11–20 years) has many genomic similarities to melanoma that occurs in adults, including an enrichment of UV-induced mutations (13). And ionizing radiation can lead to the development of leukemia and other cancers in children and adolescents. Children and adolescents who were exposed to radiation from the atomic bombs dropped in Japan during the Second World War had an elevated risk of leukemia (14), and children who were exposed to radiation from the Chernobyl nuclear plant accident had an elevated risk for thyroid cancer (15).
Exposure of parents to ionizing radiation is also a possible concern in terms of the development of cancer in their future offspring. Children whose mothers had x-rays during pregnancy (that is, children who were exposed before birth) or who were exposed after birth to diagnostic medical radiation from computed tomography (CT) scans have been found to have a slight increase in risk of leukemia and brain tumors, and possibly other cancers (16). However, genomic analysis of children born to people exposed to radiation at Chernobyl indicates that this exposure did not lead to an increase in new genetic changes being passed from parent to child (17).
Several other environmental exposures of parents have also been associated with childhood cancer. For some types of childhood leukemia (particularly acute lymphoblastic leukemia), associations have been described for father’s tobacco smoking (18, 19) and for parental exposure to certain pesticides at their workplace (20–22). Studies of childhood brain tumors have suggested associations with maternal consumption of cured meats (23).
Factors associated with reduced risks of childhood cancer
Researchers have also identified factors that may be associated with reduced risk of childhood cancer. For example, maternal consumption of folate has been associated with reduced risks of both leukemia and brain tumors in children (24). Being breastfed and having been exposed to routine childhood infections are both associated with a lower risk of developing childhood leukemia (25).
What does a child’s cancer diagnosis mean for cancer risk in the rest of the family?
First- and second-degree relatives of a child diagnosed with cancer may be at increased risk for developing cancer if there is already a family history of cancer—that is, if the child’s cancer is likely due to an inheritedgeneticdisorder (26). A clinician may advise as to whether a child and family members could benefit from genetic testing or referral to a medical geneticist or cancer genetic counselor for evaluation (26–28).
How do cancers in adolescents and young adults differ from those in younger children?
Cancer occurs more frequently in adolescents and young adults ages 15 to 39 years than in younger children, although incidence in this group is still much lower than in older adults. According to the NCI Surveillance, Epidemiology, and End Results (SEER) Program, each year in 2017–2021 there were:
17.14 cancer diagnoses per 100,000 children ages younger than 15 years
74.9 cancer diagnoses per 100,000 adolescents and young adults ages 15 to 39 years
528.9 cancer diagnoses per 100,000 adults ages 40 to 64 years
The most frequent cancers diagnosed in adolescents and young adults (AYAs) are cancers that are more common among adults than younger children, such as breast cancer, melanoma, and thyroid cancer (29). But certain cancers, such as testicular cancer, are more likely to be diagnosed among AYAs than among either younger children or adults. However, the incidence of specific cancer types varies widely across the adolescent and young adult age continuum.
Where do children and adolescents with cancer get treated?
Children and adolescents who have cancer are often treated at a children’s cancer center, which is a hospital or a unit within a hospital that specializes in diagnosing and treating patients through 20 years of age. The health professionals at these centers have specific training and expertise to provide comprehensive care for children and adolescents with cancer, and their families.
Children’s cancer centers also participate in clinical trials. The improvements in survival for children with cancer that have occurred over the past half century have been achieved because of treatment advances that were studied and proven to be effective in clinical trials.
More than 90% of children and adolescents who are diagnosed with cancer each year in the United States are cared for at a children’s cancer center that is affiliated with the NCI-supported Children’s Oncology Group (COG). COG is the world’s largest organization that performs clinical research to improve the care and treatment of children and adolescents with cancer. Each year, approximately 4,000 children who are diagnosed with cancer enroll in a COG-sponsored clinical trial. COG trials are sometimes open to older individuals when the type of cancer being studied is one that occurs in children, adolescents, and young adults.
Children’s cancer centers that participate in COG must meet and maintain strict standards of excellence for childhood cancer care. A directory of COG locations is available on their website. Families can ask their pediatrician or family doctor for a referral to a children’s cancer center. Families and health professionals can call NCI’s Cancer Information Service at 1-800-4-CANCER (1-800-422-6237) to learn more about children’s cancer centers that belong to COG.
If my child is treated at a children’s cancer center, will they automatically be part of a clinical trial?
No. Participation in a clinical trial is voluntary, and it is up to each family in collaboration with their treatment team to decide if clinical trial participation is right for their child.
Can children and adolescents who have cancer be treated at the National Institutes of Health (NIH) Clinical Center?
Children and adolescents with cancer may be eligible to be treated in clinical trials at the NIH Clinical Center in Bethesda, Maryland. Because the NIH Clinical Center is a research hospital, only patients who have a specific type or stage of cancer that is under study can be accepted for treatment. In some cases, patients with conditions that are rare or difficult to diagnose may also be accepted for treatment at the NIH Clinical Center. All patients who are treated at the NIH Clinical Center must be referred by a physician.
NCI’s Pediatric Oncology Branch conducts clinical trials at the NIH Clinical Center for children, adolescents, and young adults with a wide variety of cancers. Patients with newly diagnosed cancer, as well as patients whose cancers have come back after treatment, may be eligible to participate in a clinical trial. Physicians at the Pediatric Oncology Branch can also provide a second opinion on a patient’s diagnosis or treatment plan. To refer a patient to the Pediatric Oncology Branch, the patient’s health care provider should call 240-760-6403 (local) or 1-833-248-0479 (toll-free) weekdays between 8:30 a.m. and 5:00 p.m. ET. Parents can also call these numbers to learn if their child is eligible to participate in a clinical trial.
NCI’sClinical Genetics Branch conducts long-term observational and clinical studies of genetic susceptibility to cancer, mostly in families with a known or suspected cancer predisposition syndrome, or families that have a higher-than-expected rate of cancer. Although some of these study participants, including children and adolescents, are seen at the NIH Clinical Center, the genetic susceptibility studies do not evaluate treatment or other interventions, as in a clinical trial. Information about active studies that enroll children and adolescents can be obtained by contacting the referral nurse at 1-800-518-8474.
What should survivors of childhood and adolescent cancer consider after they complete treatment?
Some people who have had cancer during childhood or adolescence may need follow-up care and enhanced medical surveillance as they get continue to get older because of the risk of complications related to the disease or its treatment that can last for, or arise, many years after they complete treatment for their cancer (30, 31). Health problems that develop months or years after treatment has ended are known as late effects.
The specific late effects that a person who was treated for childhood cancer might experience depend on the type and location of their cancer, the type of treatment they received, and patient-related factors, such as age at diagnosis. Additionally, some people with a history of childhood cancer may need additional follow-up if an inheritedgeneticalteration is found to be the cause of the cancer.
Children and adolescents who were treated for bone cancer, brain tumors, or Hodgkin lymphoma, or who received radiation to their chest, abdomen, or pelvis, have the highest risk of serious late effects from their cancer treatment, including second cancers, joint replacement, hearing loss, and congestive heart failure (32–34).
Long-term follow-up analysis of a cohort of survivors of childhood cancer treated between 1970 and 1986 has shown that these survivors remain at risk of complications and premature death as they age, with more than half of them having experienced a severe or disabling complication or even death by the time they reach age 50 years (35). Children and adolescents treated in more recent decades may have lower risks of late effects due to modifications in treatment regimens to reduce exposure to radiotherapy and chemotherapy, increased efforts to detect late effects, and improvements in medical care for late effects (34, 36, 37).
It’s important for people who had cancer during childhood or adolescence to have regular medical follow-up examinations so any health problems can be identified and treated as soon as possible. The Children’s Oncology Group (COG) has developed long-term follow-up guidelines for survivors of childhood, adolescent, and young adult cancers.
It is also important to keep a record of the details of the cancer diagnosis and the treatment that was received. This record should include:
contact information for the hospitals and doctors who provided treatment
names and total doses of all chemotherapy drugs used in treatment
the parts of the body that were treated with radiation and the total doses of radiation that were given
types and dates of all surgeries
any other cancer treatments received
any serious complications that occurred during treatment and how those complications were treated
the date that cancer treatment was completed
The record should be kept in a safe place, and copies of the record should be given to all doctors or other health care providers who are involved with the child’s follow-up care, even as the child grows into adulthood.
Many cancer centers have programs to help patients and families navigate long-term survivor care, including both the physical and emotional issues they may face after treatment. Many children’s cancer centers have clinics where survivors of childhood cancer can go for follow-up care until they reach their early 20s. Some cancer centers are now creating clinics dedicated to follow-up care for long-term cancer survivors of pediatric and adolescent cancers. Programs focused on adolescent and young adult (AYA) cancer survivors address some of the more unique needs of these patients, including discussing and preserving future fertility, peer support, and psychosocial support that addresses personal issues such as finances, education, occupational impacts, and transition to independence.
Types of cancer in adolescents and young adults (AYAs)
An estimated 85,480 adolescents and young adults (AYAs) between the ages of 15 to 39 will be diagnosed with cancer in the United States in 2025. This accounts for about 4.2% of all cancer diagnoses.
Rates of new cancer cases of any site among AYAs have been rising on average 0.3% each year over 2013–2022, the last 10 years of available data. Death rates have been falling on average 0.9% each year over 2014–2023, according to Cancer Stat Facts: Cancer Among Adolescents and Young Adults (AYAs) (Ages 15–39), NCI’s Surveillance, Epidemiology, and End Results (SEER) Program which uses statistical models for analysis.
The most common cancers in adolescents and young adults (ages 15–39) are:
Learn how many different types of cancer are diagnosed and treated: A to Z cancer types.
Most common cancers by age group in adolescents and young adults
The incidence of specific cancer types varies according to age. Some cancers are more common in adolescents, while others are more common in young adults.
Survival rates for cancers in adolescents and young adults
Survival rates are increasing among AYAs with cancer. While relative survival rates are high, at 86% for AYAs when all cancer types are combined, survival rates are much lower for some types of cancer than for others. Learn more about survival rates for different types of cancer.
Introduction to Adolescent and Young Adult Cancers
Cancer researchers, advocates, and a cancer survivor introduce the topic of adolescent and young adult cancers.
Finding a doctor and hospital after a cancer diagnosis
When possible, it is important to get a second opinion on your cancer treatment plan. After you get a diagnosis and treatment plan from one oncologist, you may choose to get another doctor’s opinion. Most doctors will support and understand your interest in seeking a second opinion and your insurance company may even require a second opinion on the diagnosis and the treatment plan.
When possible, the second opinion should come from an oncologist who specializes in treating the type of cancer that you have. This doctor will usually be the most knowledgeable about the latest cancer treatments, including those for young adults. Seeing a specialist, and getting your initial treatment plan reviewed, can ensure that all aspects of care and treatment are considered. A second opinion may be especially helpful when the first opinion on the treatment plan comes from a doctor who doesn’t specialize in or treat many young adults with the type of cancer that you have. Learn more about finding a doctor and getting a second opinion in Finding Cancer Care.
What Adolescents and Young Adults Need to Know after a Cancer Diagnosis
Experts who treat adolescents and young adults with cancer answer the question: What would you tell an adolescent or young adult with cancer?
The type of treatment you receive is based on factors such as:
the type of cancer
how advanced the cancer is, including its stage or grade
biomarker and genetic test results for some types of cancer
your age and overall health
personal preferences
Treating cancer in adolescents and young adults
Common treatments include chemotherapy, immunotherapy, radiation therapy, stem cell transplants, surgery, and targeted therapy. Learn more about these treatments in Types of Cancer Treatment.
Your treatment options may include:
Standard medical care (also called standard of care) is treatment that experts agree is appropriate and accepted for a specific disease, such as cancer. Check out the A to Z List of Cancers to learn about treatment for a specific type of cancer.
Genetic testing for inherited cancers looks for specific inherited changes (variants) in a person’s genes. Harmful variants in some genes are known to be associated with an increased risk of developing cancer, in about 5% to 10% of all cancers.
Treatment decisions often come with questions and concerns, especially when considering participation in a clinical trial. We can help. Please contact our free, confidential Cancer Information Service at 1-800-4-CANCER (1-800-422-6237) for information and for help finding clinical trials.
Fertility preservation options
Cancer treatments can affect your fertility. It’s important to talk with your doctor about whether or not the cancer treatment being advised may increase the risk of, or cause, infertility.
Meeting with a reproductive endocrinologist can help you learn about fertility preservation options before starting treatment. Learn more about fertility issues in girls and women and fertility issues in boys and men. Research has found that although discussions of fertility preservation between doctors and young adult cancer patients are becoming more common, improvements are still needed.
Fertility Issues for Adolescents and Young Adults with Cancer
The effect of cancer treatment on fertility is a special concern for many young people with cancer.
Coping and support for young people
Cancer can create a sense of isolation from friends and family, who may not understand what you are going through. As a young person, you may feel like you are losing your independence at a time when you were just starting to gain it. Perhaps you just began college, landed a job, or started a family. A desire for normalcy may keep you from sharing your experiences with friends.
For many young people, the completion of treatment is something to celebrate. However, this time may also bring new challenges. You may worry that cancer will return or struggle to get used to new routines. Some young people enter this new phase feeling stronger, whereas others are more fragile. Most young people say the transition after treatment took longer and was more challenging than they anticipated. While most of the side effects that you had during treatment will go away, long-term side effects, such as fatigue, may take time to go away. Other side effects, called late effects, may not occur until months or even years after treatment.
Although follow-up care is important for all survivors, it is especially important for adolescents and young adults. These check-ups can both reassure you and help to prevent or treat medical and psychological problems. Some young people receive follow-up care at the hospital where they were treated, and others see specialists at late effect or survivorship care clinics. Talk with your health care team to learn what follow-up care you should receive and about possible places to receive this important care. Learn more in questions to ask your doctor when you have finished cancer treatment.
Important documents to get from your doctor and talk about include:
Treatment summary, with detailed records about your diagnosis and the type(s) of treatment you received.
Survivorship care plan or follow-up care plan, which addresses both physical and psychological follow-up care that you should receive after cancer treatment. The plan is based on the type of cancer and treatment you received and may vary from person to person.
You’ll want to save both paper and electronic copies of these documents for your medical records. Learn more about Follow-Up Medical Care.
Cancer survivorship clinics and support groups
A growing number of hospitals have survivorship clinics, including programs that focus on follow-up care for young people who have been treated for cancer. Research is also ongoing through the Childhood Cancer Survivor Study to both increase survival rates and to better understand and minimize late effects.
If the cancer you have cannot be cured, or continues to progress despite treatment, it may be called end-stage cancer or terminal cancer. Being told that treatment isn’t working is exceptionally difficult to hear. Get suggestions that can help you have conversations and make choices about end-of-life care and support that are right for you: Young People Facing End-of-Life Care Decisions.
Organizations serving adolescents and young adults
A growing number of organizations serve the needs of AYAs with cancer. Some organizations help young people connect with peers who are going through the same things. Others address topics such as coping, fertility, and survivorship.
Late effects are health problems that occur months or years after cancer treatment has ended.
Late effects in childhood cancer survivors affect the body and mind.
There are three important factors that affect the risk of late effects.
The chance of having late effects increases over time.
Regular follow-up care is very important for survivors of childhood cancer.
Good health habits are also important for survivors of childhood cancer.
Late effects are health problems that occur months or years after cancer treatment has ended.
Cancer treatments may harm the body’s organs, tissues, or bones and cause health problems later in life. They may include physical, mental, and social problems and second cancers. These health problems are called late effects.
Doctors are studying the late effects caused by cancer treatment. They are working to improve cancer treatments and stop or lessen late effects. While most late effects are not life-threatening, they may cause serious problems that affect health and quality of life.
Late effects in childhood cancer survivors affect the body and mind.
Late effects in childhood cancer survivors may affect:
There are three important factors that affect the risk of late effects.
Many childhood cancer survivors will have late effects. The risk of late effects depends on factors related to the tumor, treatment, and patient. These include:
Cancer-related factors
type of cancer
the organs and tissues that were affected by the cancer
Treatment-related factors
type of radiation therapy, part of the body treated, and dose
The chance of having late effects increases over time.
New treatments for childhood cancer have decreased the number of deaths from the primary cancer. Because childhood cancer survivors are living longer, they are having more late effects after cancer treatment. Survivors may not live as long as people who did not have cancer. The most common causes of death in childhood cancer survivors are:
Studies of the causes of late effects have led to changes in treatment. This has improved the quality of life for cancer survivors and helps prevent illness and death from late effects.
Regular follow-up care is very important for survivors of childhood cancer.
Regular follow-up by health professionals who are trained to find and treat late effects is important for the long-term health of childhood cancer survivors. Follow-up care will be different for each person who has been treated for cancer. The care plan the cancer survivor receives will depend on their general health and health habits, the type of cancer, the type of treatment, genetic factors, and other medical conditions. Follow-up care includes checking for symptoms of late effects and health education on how to prevent or lessen late effects.
It is important that childhood cancer survivors have an exam at least once a year. The exams should be done by a health professional who knows the survivor’s risk for late effects and can recognize the early signs of late effects. Blood and imaging tests may also be done.
Long-term follow-up may improve the health and quality of life for cancer survivors. It also helps doctors study the late effects of cancer treatments so that safer therapies for newly diagnosed children may be developed.
Good health habits are also important for survivors of childhood cancer.
The quality of life for cancer survivors may be improved by behaviors that promote health and well-being. These include a healthy diet, exercise, and regular medical and dental check-ups. These self-care behaviors are especially important for cancer survivors because of their risk of health problems related to treatment. Healthy behaviors may make late effects less severe and lower the risk of other diseases.
Avoiding behaviors that are damaging to health is also important. Smoking, excess alcohol use, illegal drug use, not following good sun protection measures, or not being physically active may worsen organ damage related to treatment and may increase the risk of second cancers.
Second Cancers
Key Points
Childhood cancer survivors have an increased risk of a second cancer later in life.
Certain genetic patterns or syndromes may increase the risk of a second cancer.
People who have been treated for cancer need regular screening tests to check for a second cancer.
The kind of test used to screen for a second cancer depends in part on the kind of cancer treatment the survivors had in the past.
Childhood cancer survivors have an increased risk of a second cancer later in life.
A different primary cancer that occurs at least two months after cancer treatment ends is called a second cancer. A second cancer may occur months or years after treatment is completed. The type of second cancer that occurs depends in part on the original type of cancer and the cancer treatment. Benign tumors (not cancer) may also occur.
Second cancers that may occur after cancer treatment include:
Children treated with radiation to the bone marrow without chemotherapy may also have an increased risk of second acute myeloid leukemia.
Solid tumors that may appear more than 10 years after a primary cancer diagnosis and treatment include:
Breast cancer. Survivors of childhood, adolescent, and young adult cancers who received high-dose radiation therapy to the chest have an increased risk of developing breast cancer. Certain chemotherapy drugs, including alkylating agents and anthracyclines, also increase a person’s risk of breast cancer. Risk is highest for female survivors of Hodgkin lymphoma, sarcoma, and leukemia.
Brain tumors. Brain tumors may occur after radiation therapy to the head and/or intrathecal chemotherapy using methotrexate for a primary brain tumor or for cancer that has spread to the brain or spinal cord, such as acute lymphocytic leukemia or non-Hodgkin lymphoma. When intrathecal chemotherapy using methotrexate and radiation treatment are given together, the risk of a brain tumor is even higher.
Bone and soft tissue tumors. There is an increased risk of bone and soft tissue tumors after radiation treatment for hereditaryretinoblastoma, Ewing sarcoma, and other cancers of the bone, with risks increasing at higher doses. Chemotherapy with anthracyclines or alkylating agents also increases the risk of bone and soft tissue tumors, with risk increasing at higher doses.
Lung cancer. There is an increased risk of lung cancer after radiation treatment to the chest for Hodgkin lymphoma or bone cancer, especially in patients who smoke.
Stomach, liver, or colorectal cancer. Stomach, liver, or colorectal cancer may occur after receiving radiation therapy to the abdomen or pelvis, chemotherapy, or both. Survivors of Wilms tumor and Hodgkin lymphoma have the highest risk of these second cancers. The risk increases with higher doses of radiation. There is also an increased risk of colorectalpolyps.
Nonmelanoma skin cancer (basal cell carcinoma or squamous cell carcinoma). There is an increased risk of nonmelanoma skin cancer after radiation treatment, and it usually appears in the area where radiation was given. Being exposed to UV radiation may increase this risk. Patients who develop nonmelanoma skin cancer after radiation treatment have an increased chance of developing other types of cancers in the future. The risk of basal cell carcinoma is also increased after treatment with chemotherapy drugs, such as vincristine and vinblastine (called vinca alkaloids). Survivors of hereditary retinoblastoma may have an increased risk of developing nonmelanoma skin cancer.
Malignant melanoma. Malignant melanoma may occur after radiation or combination chemotherapy with alkylating agents and antimitotic drugs (such as vincristine and vinblastine). Survivors of Hodgkin lymphoma, hereditary retinoblastoma, soft tissue sarcoma, and gonadal tumors are more likely to be at a higher risk of having malignant melanoma. Malignant melanoma as a second cancer is less common than nonmelanoma skin cancer. Survivors of hereditary retinoblastoma may have an increased risk of developing melanoma.
Oral, laryngeal, and salivary gland cancer. Oral cancer may occur after stem cell transplant with a history of chronic graft-versus-host disease, radiation therapy, or chemotherapy. Survivors of leukemia, osteosarcoma, Hodgkin lymphoma, and soft tissue sarcoma have the highest risk of oral cancers. Childhood, adolescent, and young adult cancer survivors may also be at increased risk of developing oral cancers related to infection with human papillomavirus.
Kidney cancer. There is an increased risk of kidney cancer after treatment for neuroblastoma, radiation treatment to the middle of the back, or chemotherapy such as cisplatin or carboplatin.
Bladder cancer and genital cancer. Bladder cancer may occur after chemotherapy with cyclophosphamide. Survivors of heritable retinoblastoma have an increased risk of developing bladder cancer. Female survivors have an increased risk of cancer in the uterus or vulva.
Certain genetic patterns or syndromes may increase the risk of a second cancer.
Some childhood cancer survivors may have an increased risk of developing a second cancer because they have a family history of cancer, an inherited cancer syndrome such as Li-Fraumeni syndrome, or a geneticmutation such as neurofibromatosis type 1. Problems with the way DNA is repaired in cells and the way anticancer drugs are used by the body may also affect the risk of second cancers.
People who have been treated for cancer need regular screening tests to check for a second cancer.
It is important for people who have been treated for cancer to be checked for a second cancer before symptoms appear. This is called screening and may help find a second cancer when it is smaller and has not spread. When abnormal tissue or cancer is found early, it may be easier to treat. By the time symptoms appear, cancer may have begun to spread.
It is important to remember that the doctor does not necessarily think a childhood cancer survivor has cancer if he or she suggests a screening test. Screening tests are given when the cancer survivor has no cancer symptoms. If a screening test result is abnormal, more tests may be done to find out if there is a second cancer. These are called diagnostic tests.
The kind of test used to screen for a second cancer depends in part on the kind of cancer treatment the survivors had in the past.
Childhood cancer survivors should have a physical exam and medical history done once a year. A physical exam of the body is done to see how healthy the person is and to check for signs of disease, such as lumps, changes in the skin, or anything else that seems unusual. A medical history is taken to learn about the cancer survivor’s health habits and past illnesses and treatments.
Childhood cancer survivors who received radiation therapy may have the following tests and procedures to check for skin, breast, or colorectal cancer:
Skin exam is when a doctor or nurse checks the skin for bumps or spots that look abnormal in color, size, shape, or texture, especially in the area where radiation was given. It is suggested that a skin exam be done once a year to check for signs of skin cancer. The doctor will also provide information about sun protection behaviors to reduce the risk of skin cancer.
Breast self-exam is an exam of the breast by the patient. The patient carefully feels the breasts and under the arms for lumps or anything else that seems unusual. It is suggested that women treated with a higher dose of radiation therapy to the chest do a monthly breast self-exam beginning at puberty. Talk to the doctor about when breast self-exams should begin.
Clinical breast exam (CBE) is an exam of the breast by a doctor or other health professional. The doctor will carefully feel the breasts and under the arms for lumps or anything else that seems unusual. It is suggested that women treated with a higher dose of radiation therapy to the chest have a clinical breast exam every year beginning at puberty until age 25. After age 25 or 8 years after radiation treatments end (whichever is first), clinical breast exams are done every 6 months. Talk to the doctor about when clinical breast exams should begin.
Mammogram is an x-ray of the breast. A mammogram may be done in women who had a higher dose of radiation to the chest and who do not have dense breasts. It is suggested that these women have a mammogram once a year starting 8 years after treatment or at age 25, whichever is later. Talk to the doctor about when mammograms to check for breast cancer should begin.
Breast MRI (magnetic resonance imaging) uses a magnet, radio waves, and a computer to make a series of detailed pictures of the breast. This procedure is also called nuclear magnetic resonance imaging (NMRI). An MRI may be done in women who had a higher dose of radiation to the chest and who have dense breasts. It is suggested that these women have an MRI once a year starting 8 years after treatment or at age 25, whichever is later. Talk to the doctor about whether an MRI of the breast to check for breast cancer is needed.
Colonoscopy is a procedure to look inside the rectum and colon for polyps, abnormal areas, or cancer. A colonoscope is inserted through the rectum into the colon. A colonoscope is a thin, tube-like instrument with a light and a lens for viewing. It may also have a tool to remove polyps or tissue samples, which are checked under a microscope for signs of cancer. It is suggested that childhood cancer survivors who had a higher dose of radiation to the abdomen, pelvis, or spine have a colonoscopy every 5 years. This begins at age 30 or 5 years after treatment ended, whichever is later. Talk to your doctor about when colonoscopies to check for colorectal cancer should begin.
Multitarget stoolDNA test is a test to collect stool to check for cancer cells. This test may be done every 3 years beginning at age 30 or 5 years after treatment ended.
Cardiovascular System
Key Points
Heart and blood vessel late effects are more likely to occur after treatment for certain childhood cancers.
Radiation to the chest and certain types of chemotherapy increase the risk of heart and blood vessel late effects.
Late effects that affect the heart and blood vessels may cause certain health problems.
Possible symptoms of heart and blood vessel late effects include trouble breathing and chest pain.
Certain tests and procedures are used to diagnose health problems in the heart and blood vessels.
Health habits that promote a healthy heart and blood vessels are important for survivors of childhood cancer.
Heart and blood vessel late effects are more likely to occur after treatment for certain childhood cancers.
Cardiovascular disease occurs at a younger age in childhood cancer survivors. Treatment for these and other childhood cancers may cause heart and blood vessel late effects:
Radiation to the chest and certain types of chemotherapy increase the risk of heart and blood vessel late effects.
The risk of health problems involving the heart and blood vessels increases after treatment with:
Radiation to the chest, spine, brain, neck, kidneys, or total-body irradiation (TBI) as part of a stem cell transplant. The risk of problems depends on the area of the body that was exposed to radiation, the amount of radiation given, and whether the radiation was given in small or large doses.
Certain types of chemotherapy. Chemotherapy with anthracyclines such as doxorubicin, daunorubicin, idarubicin, and epirubicin, and with anthraquinones such as mitoxantrone increase the risk of heart and blood vessel problems. The risk of problems depends on the total dose of chemotherapy given and the type of drug used. It also depends on whether treatment with anthracyclines was given to a child younger than 13 years and whether a drug called dexrazoxane was given during treatment with anthracyclines. Dexrazoxane may lessen heart and blood vessel damage up to 5 years after treatment. Ifosfamide, cyclophosphamide, methotrexate, asparaginase, and chemotherapy with a platinum, such as carboplatin and cisplatin, may also cause heart and blood vessel late effects.
Stem cell transplant.
Nephrectomy (surgery to remove all or part of a kidney).
Childhood cancer survivors who were treated with both radiation therapy to the heart or blood vessels and certain types of chemotherapy are at greatest risk.
Other factors that may also increase the risk of heart and blood vessel late effects include:
Longer time since treatment.
Having high blood pressure or other risk factors for heart disease, such as a family history of heart disease, having excess body weight, smoking, high cholesterol, or diabetes. When these risk factors are combined, the risk of late effects is even higher.
New treatments that decrease the amount of radiation given, use lower doses of chemotherapy or less harmful chemotherapy drugs, and incorporate dexrazoxane may be leading to a lower risk of heart and blood vessel late effects in childhood cancer survivors.
Late effects that affect the heart and blood vessels may cause certain health problems.
Childhood cancer survivors who received radiation or certain types of chemotherapy have an increased risk of late effects to the heart and blood vessels and related health problems. These include:
when exposed to cold or having strong emotions, the fingers, toes, ears, or nose become white and then turn blue, and sometimes the fingers will also have pain or tingling
sudden numbness or weakness of the face, arm, or leg (especially on one side of the body)
sudden confusion or trouble speaking or understanding speech
sudden trouble seeing with one or both eyes
sudden trouble walking or feeling dizzy
sudden loss of balance or coordination
sudden severe headache for no known reason
pain, warmth, or redness in one area of the arm or leg, especially the back of the lower leg
The only way to know if these symptoms are caused by heart and blood vessel late effects is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the heart and blood vessels.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Electrocardiogram (EKG) is a recording of the heart’s electrical activity to check its rate and rhythm. A number of small pads (electrodes) are placed on the patient’s chest, arms, and legs, and are connected by wires to the EKG machine. Heart activity is then recorded as a line graph on paper. Electrical activity that is faster or slower than normal may be a sign of heart disease or damage.
Echocardiogram is a procedure in which high-energy sound waves (ultrasound) are bounced off the heart and nearby tissues or organs and make echoes. A moving picture is made of the heart and heart valves as blood is pumped through the heart.
Ultrasound exam uses high-energy sound waves (ultrasound) that bounce off internal tissues or organs such as the heart and make echoes. The echoes form a picture of body tissues called a sonogram.
Magnetic resonance imaging (MRI) uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the body. This procedure is also called nuclear magnetic resonance imaging (NMRI). This procedure is done to check for blood clots.
CT scan (CAT scan) uses a computer linked to an x-ray machine to make a series of detailed pictures of areas inside the body. The pictures are taken from different angles and are used to create 3-D views of tissues and organs. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. This procedure is done to check for blood clots. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography. Learn more about Computed Tomography (CT) Scans and Cancer.
Magnetic resonance angiography (MRA) uses radio waves and a powerful magnet linked to a computer to create detailed pictures of the blood vessels and blood flow inside the body. A dye may be injected into a vein to make the blood vessels and blood flow easier to see. This may be used to check for blood clots or other blood vessel problems.
Lipid profile studies use a blood sample to measure the amounts of triglycerides, cholesterol, and low- and high-density lipoprotein cholesterol in the blood.
Talk to the doctor about whether tests and procedures are needed to check for signs of heart and blood vessel late effects. If tests are needed, find out how often they should be done.
Health habits that promote a healthy heart and blood vessels are important for survivors of childhood cancer.
Childhood cancer survivors may lower their risk of heart and blood vessel late effects by having a healthy lifestyle, which includes:
a healthy weight
a heart-healthy diet, high in vegetables, fruits, whole grains, fish, and poultry, and low in red or processed meats
regular exercise
not smoking
Central Nervous System
Key Points
Brain and spinal cord late effects are more likely to occur after treatment of brain tumors and acute lymphoblastic leukemia.
Radiation to the brain increases the risk of brain and spinal cord late effects.
Late effects that affect the brain and spinal cord may cause certain health problems.
Possible symptoms of brain and spinal cord late effects include headaches, loss of coordination, and seizures.
Certain tests and procedures are used to diagnose health problems in the brain and spinal cord.
Survivors of childhood cancer have an increased risk of mental health disorders.
Some childhood cancer survivors have post-traumatic stress disorder.
Adolescents who are diagnosed with cancer may have social problems later in life.
Brain and spinal cord late effects are more likely to occur after treatment of brain tumors and acute lymphoblastic leukemia.
Treatment for these and other childhood cancers may cause brain and spinal cordlate effects:
Late effects that affect the brain and spinal cord may cause certain health problems.
Childhood cancer survivors who received radiation, certain types of chemotherapy, or surgery to the brain or spinal cord have an increased risk of late effects to the brain and spinal cord and related health problems. A child may develop these problems soon after treatment or years later in adulthood. Brain and spinal cord late effects include:
headaches that may go away after vomiting
loss of balance, lack of coordination, or trouble walking
movement disorders that affect the legs and eyes or the ability to speak and swallow
nerve damage in the hands or feet
stroke, with an increased risk for a second stroke for survivors who received radiation to the brain, have a history of high blood pressure, or were older than 40 years when they had their first stroke
a small amount of bleeding in the brain (brain microbleeds)
social withdrawal or trouble getting along with others
The only way to know if these symptoms are caused by brain and spinal cord late effects is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the brain and spinal cord.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Neurological exam is a series of questions and tests to check the brain, spinal cord, and nerve function. The exam checks a person’s mental status, coordination, and ability to walk normally, and how well the muscles, senses, and reflexes work. This may also be called a neuro exam or a neurologic exam. In some cases, a more complete exam may be done by a neurologist or neurosurgeon.
Neuropsychological assessment is a series of tests to examine the patient’s mental processes and behavior. Areas that are checked usually include:
knowing who and where you are and what day it is
ability to learn and remember new information
intelligence
ability to solve problems
use of spoken and written language
eye-hand coordination
ability to organize information and tasks
Talk to the doctor about whether tests and procedures are needed to check for signs of brain and spinal cord late effects. If tests are needed, find out how often they should be done.
Survivors of childhood cancer have an increased risk of mental health disorders.
Survivors of childhood cancer are more likely to be diagnosed with mental health disorders than their peers who have not had cancer. These disorders include:
Having a mental health disorder may cause problems with personal relationships, education, employment, and health, and cause thoughts of suicide. As a result, survivors may be less likely to live on their own as adults.
Follow-up care for childhood cancer survivors should include screening and treatment for possible psychologicaldistress or mental health disorders.
For more information about psychological distress and coping skills for people with cancer, see:
Some childhood cancer survivors have post-traumatic stress disorder.
Being diagnosed and treated for a life-threatening disease may be traumatic. This trauma may cause post-traumatic stress disorder (PTSD). PTSD is defined as having certain behaviors following a stressful event that involved death or the threat of death, serious injury, or a threat to oneself or others.
PTSD can affect cancer survivors in the following ways:
constantly reliving or thinking about the time they were diagnosed and treated for cancer
having nightmares or flashbacks
avoiding places, events, and people that remind them of the cancer experience
In general, childhood cancer survivors show low levels of PTSD, depending in part on their coping style and the coping style of their parents. Survivors who received radiation therapy to the head when younger than 4 years or survivors who received intensive treatment may be at higher risk of PTSD. Family problems, little or no social support from family or friends, and stress not related to the cancer may increase the chances of having PTSD.
Because avoiding places and people connected to the cancer may be part of PTSD, survivors with PTSD may not get the medical treatment they need.
Adolescents who are diagnosed with cancer may have social problems later in life.
Adolescents who are diagnosed with cancer may reach fewer social milestones or reach them later in life than adolescents not diagnosed with cancer. Social milestones include having a first boyfriend or girlfriend, getting married, and having a child. They may also have trouble getting along with other people or feel like they are not liked by others their age.
Cancer survivors in this age group have reported being less satisfied with their health and their lives in general compared with others of the same age who did not have cancer. Adolescents and young adults who survived cancer need special programs that give psychological, educational, and job support.
Digestive System
Key Points
Teeth and jaws
Problems with the teeth and jaws are more likely to occur after treatment for certain childhood cancers.
Radiation to the head and neck and certain types of chemotherapy increase the risk of late effects to the teeth and jaws.
Late effects that affect the teeth and jaws may cause certain health problems.
Possible symptoms of late effects of the teeth and jaws include tooth decay (cavities) and jaw pain.
Certain tests and procedures are used to diagnose health problems in the mouth and jaws.
Regular dental care is very important for survivors of childhood cancer.
Digestive tract
Digestive tract late effects are more likely to occur after treatment for certain childhood cancers.
Radiation to the bladder, prostate, or testicles and certain types of chemotherapy increase the risk of digestive tract late effects.
Late effects that affect the digestive tract may cause certain health problems.
Possible symptoms of digestive tract late effects include abdominal pain and diarrhea.
Certain tests and procedures are used to diagnose health problems in the digestive tract.
Liver and bile ducts
Liver and bile duct late effects are more likely to occur after treatment for certain childhood cancers.
Certain types of chemotherapy and radiation to the liver or bile ducts increase the risk of late effects.
Late effects that affect the liver and bile ducts may cause certain health problems.
Possible symptoms of liver and bile duct late effects include abdominal pain and jaundice.
Certain tests and procedures are used to diagnose health problems in the liver and bile duct.
Health habits that promote a healthy liver are important for survivors of childhood cancer.
Pancreas
Radiation therapy increases the risk of pancreatic late effects.
Late effects that affect the pancreas may cause certain health problems.
Possible symptoms of pancreatic late effects include frequent urination and being thirsty.
Certain tests and procedures are used to diagnose health problems in the pancreas.
Treatment with chemotherapy, radiation therapy, or surgery may cause problems throughout the digestive system, which starts at the mouth and ends at the rectum.
Teeth and jaws
Problems with the teeth and jaws are more likely to occur after treatment for certain childhood cancers.
Treatment for these and other childhood cancers may cause late effects of the teeth and jaws:
Possible symptoms of late effects of the teeth and jaws include tooth decay (cavities) and jaw pain.
These and other symptoms may be caused by late effects of the teeth and jaws or by other problems:
teeth are small or do not have a normal shape
missing permanent teeth
permanent teeth come in at a later than normal age
teeth have less enamel than normal
more tooth decay (cavities) and gum disease than normal
dry mouth
trouble chewing, swallowing, and speaking
jaw pain
jaws do not open and close the way they should
The only way to know if these symptoms are caused by late effects of the teeth and jaws is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the mouth and jaws.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Dental exam is an exam of the teeth, mouth, and jaws to check general signs of dental health, including checking for signs of disease, such as cavities or anything that seems unusual. This may also be called a dental check-up.
Panorex x-ray is an x-ray of all of the teeth and their roots. An x-ray is a type of radiation that can go through the body and make pictures.
X-ray of the jaws is an x-ray of the jaws. An x-ray is a type of radiation that can go through the body and make pictures.
CT scan (CAT scan) uses a computer linked to an x-ray machine to make a series of detailed pictures of areas inside the body, such as the head and neck. The pictures are taken from different angles and are used to create 3-D views of tissues and organs. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography. Learn more about Computed Tomography (CT) Scans and Cancer.
Magnetic resonance imaging (MRI) uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the body, such as the head and neck. This procedure is also called nuclear magnetic resonance imaging (NMRI).
Biopsy is a procedure in which a sample of tissue is removed from the tumor so that a pathologist can view it under a microscope to check for signs of cancer.
Talk to the doctor about whether tests and procedures are needed to check for signs of teeth and jaw late effects. If tests are needed, find out how often they should be done.
Regular dental care is very important for survivors of childhood cancer.
Doctors suggest that survivors of childhood cancer have a dental check-up and a cleaning and fluoride treatment every 6 months. Children who had radiation therapy to the oral cavity may also see an orthodontist or an otolaryngologist. If lesions are present in the mouth, a biopsy may be needed.
Digestive tract
Digestive tract late effects are more likely to occur after treatment for certain childhood cancers.
Radiation to the bladder, prostate, or testicles and certain types of chemotherapy increase the risk of digestive tract late effects.
The risk of health problems that affect the digestive tract increases after treatment with:
Radiation therapy to the abdomen or areas near the abdomen, such as the esophagus, bladder, prostate, or testicles, may cause digestive tract problems that begin quickly and last for a short time. In some patients, however, digestive tract problems are delayed and long-lasting. These late effects are caused by radiation therapy that damages the blood vessels. Receiving higher doses of radiation therapy or receiving chemotherapy such as dactinomycin or anthracyclines together with radiation therapy may increase this risk.
The only way to know if these symptoms are caused by digestive late effects is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the digestive tract.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Digital rectal exam is an exam where the doctor or nurse inserts a lubricated, gloved finger into the rectum to feel for lumps or anything else that seems unusual.
Blood chemistry studies is a test that uses a blood sample to measure the amounts of certain substances released into the blood by organs and tissues in the body. An unusual amount of a substance can be a sign of disease.
Kidney, ureter, and bladder x-ray is a type of radiation that can go through the body and make pictures of the abdomen, kidney, ureter, or bladder to check for signs of disease.
CT scan (CAT scan) uses a computer linked to an x-ray machine to make a series of detailed pictures of areas inside the body. The pictures are taken from different angles and are used to create 3-D views of tissues and organs. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography. Learn more about Computed Tomography (CT) Scans and Cancer.
Magnetic resonance imaging (MRI) uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the body. This procedure is also called nuclear magnetic resonance imaging (NMRI).
Barium swallow is a series of x-rays of the throat, esophagus, stomach, and the first part of the small intestine (duodenum). For this procedure, the patient drinks a liquid that contains barium (a silver-white metallic compound). The barium coats the esophagus and stomach which helps them show up more clearly in x-rays. This procedure is also called an upper GI series.
Talk to the doctor about whether tests and procedures are needed to check for signs of digestive tract late effects. If tests are needed, find out how often they should be done.
Liver and bile ducts
Liver and bile duct late effects are more likely to occur after treatment for certain childhood cancers.
Treatment for these and other childhood cancers may cause liver or bile duct late effects:
The only way to know if these symptoms are caused by liver and bile duct late effects is to see a doctor.
Sometimes there are no signs or symptoms of liver or bile duct late effects and treatment may not be needed.
Certain tests and procedures are used to diagnose health problems in the liver and bile duct.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Blood chemistry studies use a blood sample to measure the amounts of certain substances released into the blood by organs and tissues in the body. An unusual amount of a substance can be a sign of disease. For example, there may be a higher level of bilirubin, alanine aminotransferase (ALT), and aspartate aminotransferase (AST) in the body if the liver has been damaged.
Ferritin level uses a blood sample to measure the amount of ferritin. Ferritin is a protein that binds to iron and stores it for use by the body. After a stem cell transplant, a high ferritin level may be a sign of liver disease.
the portion of the blood sample made up of red blood cells
This test is used to check the amount of platelets in the body.
Prothrombin time (PT) test measures how long it takes for the blood to clot.
Hepatitis assay uses a blood sample to check for pieces of the hepatitis virus. The blood sample may also be used to measure how much hepatitis virus is in the blood. All patients who had a blood transfusion before 1972 should have a screening test for hepatitis B. Patients who had a blood transfusion before 1993 should have a screening test for hepatitis C.
Ultrasound exam uses high-energy sound waves (ultrasound) that bounce off internal tissues or organs, such as the gall bladder, and make echoes. The echoes form a picture of body tissues called a sonogram.
CT scan (CAT scan) uses a computer linked to an x-ray machine to make a series of detailed pictures of areas inside the body. The pictures are taken from different angles and are used to create 3-D views of tissues and organs. A dye may be injected into a vein or swallowed to help the organs and tissue show up more clearly. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography. Learn more about Computed Tomography (CT) Scans and Cancer.
Magnetic resonance imaging (MRI) uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the body. This procedure is also called nuclear magnetic resonance imaging (NMRI).
Talk to the doctor about whether tests and procedures are needed to check for signs of liver or bile duct late effects. If tests are needed, find out how often they should be done.
Health habits that promote a healthy liver are important for survivors of childhood cancer.
Childhood cancer survivors with liver late effects should take care to protect their health, including:
having a healthy weight
not drinking alcohol
getting vaccines for hepatitis A and hepatitis B viruses
Pancreas
Radiation therapy increases the risk of pancreatic late effects.
The risk of pancreatic late effects may be increased in childhood cancer survivors after treatment with:
radiation therapy to the abdomen
total-body irradiation as part of a stem cell transplant
Late effects that affect the pancreas may cause certain health problems.
Pancreatic late effects and related health problems include:
Insulin resistance is a condition in which the body does not use insulin the way it should. Insulin is needed to help control the amount of glucose (a type of sugar) in the body. Because the insulin does not work the way it should, glucose and fat levels rise.
Diabetes mellitus is a disease in which the body does not make enough insulin or does not use it the way it should. When there is not enough insulin, the amount of glucose in the blood increases and the kidneys make a large amount of urine. Survivors also have an increased risk of prediabetes, which can lead to diabetes.
Possible symptoms of pancreatic late effects include frequent urination and being thirsty.
These and other symptoms may be caused by pancreatic late effects or by other problems:
frequent urination
feeling very thirsty
feeling very hungry
weight loss for no known reason
feeling very tired
frequent infections, especially of the skin, gums, or bladder
blurred vision
cuts or bruises that are slow to heal
numbness or tingling in the hands or feet
The only way to know if these symptoms are caused by pancreatic late effects is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the pancreas.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Glycated hemoglobin (A1C) test uses a sample of blood to measure the amount of glucose that is attached to red blood cells. A higher-than-normal amount of glucose attached to red blood cells can be a sign of diabetes mellitus.
Fasting blood sugar test uses a blood sample to measure the amount of glucose in the blood. This test is done after the patient has had nothing to eat overnight. A higher-than-normal amount of glucose in the blood can be a sign of diabetes mellitus.
Endocrine System
Key Points
Thyroid gland
Thyroid late effects are more likely to occur after treatment for certain childhood cancers.
Radiation therapy to the head and neck increases the risk of thyroid late effects.
Late effects that affect the thyroid may cause certain health problems.
Symptoms of thyroid late effects depend on whether there is too little or too much thyroid hormone in the body.
Certain tests and procedures are used to diagnose health problems in the thyroid.
Hypothalamus and pituitary gland
Neuroendocrine late effects may be caused after treatment for certain childhood cancers.
Treatment that affects the hypothalamus or pituitary gland increases the risk of neuroendocrine system late effects.
Late effects that affect the hypothalamus may cause certain health problems.
Certain tests and procedures are used to diagnose health problems in the neuroendocrine system.
Metabolic syndrome
Metabolic syndrome is more likely to occur after treatment for certain childhood cancers.
Radiation therapy increases the risk of metabolic syndrome.
Certain tests and procedures are used to diagnose metabolic syndrome.
Metabolic syndrome may cause heart and blood vessel disease and diabetes.
Weight changes and frailty
Weight changes and frailty are late effects that are more likely to occur after treatment for certain childhood cancers.
Radiation therapy increases the risk of weight changes and frailty.
Certain tests and procedures are used to diagnose a change in weight.
Thyroid gland
Thyroid late effects are more likely to occur after treatment for certain childhood cancers.
Treatment for these and other childhood cancers may cause thyroid late effects:
The risk also is increased in females, in survivors who were a young age at the time of treatment, in survivors who had a higher radiation dose, and as the time since diagnosis and treatment gets longer. Risk for endocrine problems also depends on the location of the tumor.
Late effects that affect the thyroid may cause certain health problems.
Thyroid late effects and related health problems include:
Hypothyroidism (not enough thyroid hormone): There is a higher risk of hypothyroidism in survivors treated with head and neck radiation involving the thyroid gland, especially survivors of Hodgkin lymphoma. This is the most common thyroid late effect. It usually occurs 2 to 5 years after treatment ends but may occur more than 25 years after radiation therapy. It is more common in girls than boys. Children, adolescents, and young adults treated with proton beam radiation therapy may have a lower risk of developing hypothyroidism.
Hyperthyroidism (too much thyroid hormone): The risk of hyperthyroidism increases with higher doses of radiation therapy to the thyroid gland. Hyperthyroidism is less common than hypothyroidism. It usually occurs 5 years after treatment ends, but may occur more than 25 years after radiation therapy.
Lumps in the thyroid: Higher radiation dose and longer time since diagnosis are linked to an increased risk of developing thyroid lumps. These growths may be benign (not cancerous) or malignant (cancer). It is more common in girls than boys. An enlarged thyroid (goiter) may be caused by lumps in the thyroid.
Symptoms of thyroid late effects depend on whether there is too little or too much thyroid hormone in the body.
These and other symptoms may be caused by thyroid late effects or by other problems:
The only way to know if these symptoms are caused by thyroid late effects is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the thyroid.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Blood hormone studies use a blood sample to measure the amounts of certain hormones released into the blood by organs and tissues in the body. An unusual amount of a substance can be a sign of disease in the organ or tissue that makes it. The blood may be checked for abnormal levels of thyroid-stimulating hormone (TSH) or free thyroxine (T4).
Ultrasound exam uses high-energy sound waves (ultrasound) that bounce off internal tissues or organs and make echoes. The echoes form a picture of body tissues called a sonogram. This procedure can show the size of the thyroid and whether there are nodules (lumps) on the thyroid.
Talk to the doctor about whether tests and procedures are needed to check for signs of thyroid late effects. If tests are needed, find out how often they should be done.
Hypothalamus and pituitary gland
Neuroendocrine late effects may be caused after treatment for certain childhood cancers.
Treatment that affects the hypothalamus or pituitary gland increases the risk of neuroendocrine system late effects.
Childhood cancer survivors have an increased risk for neuroendocrine late effects. These effects are caused by radiation therapy to treat cancer in the brain near the hypothalamus or used as total-body irradiation before a stem cell transplant. The hypothalamus controls the way hormones are made and released into the bloodstream by the pituitary gland. These effects are also caused by surgery in the area of the hypothalamus, pituitary gland, or optic pathways.
Late effects that affect the hypothalamus may cause certain health problems.
Childhood cancer survivors who have neuroendocrine late effects may have abnormal levels of hormones made in the pituitary gland and released into the blood. Neuroendocrine late effects and related health problems include:
Growth hormone deficiency: Growth hormone helps promote growth and control metabolism. A low level of growth hormone is a common late effect of radiation to the brain in childhood cancer survivors. The higher the radiation dose and the longer the time since treatment, the greater the risk of this late effect. A low level of growth hormone may also occur in childhood ALL and stem cell transplant survivors who received radiation therapy to the brain and spinal cord and/or chemotherapy.
A low level of growth hormone in childhood results in adult height that is shorter than normal. If the child’s bones have not fully developed, low growth hormone levels may be treated with growth hormone replacement therapy beginning one year after the end of treatment.
Adrenocorticotropin deficiency (ACTH): Adrenocorticotropic hormone controls the making of glucocorticoids. A low level of adrenocorticotropic hormone is an uncommon late effect. It may occur in survivors:
of childhood brain tumors, other tumors, or blood cancers
with low growth hormone levels or central hypothyroidism
who received radiation therapy to the brain
who received steroid hormone therapy
Symptoms of adrenocorticotropin deficiency may not be severe and may not be noticed. Symptoms include:
Low levels of adrenocorticotropin may be treated with hydrocortisone therapy.
Hyperprolactinemia: Prolactin controls the making of breast milk and has many other effects in the body. A high level of the hormone prolactin may occur after a high dose of radiation to the brain or surgery that affects part of the pituitary gland. A high level of prolactin may cause:
Sometimes there are no signs and symptoms. Treatment is rarely needed.
Thyroid-stimulating hormone (TSH) deficiency: Thyroid-stimulating hormone controls the making of thyroid hormones. A low level of thyroid hormone may occur very slowly over time after radiation therapy to the brain. Thyroid-stimulating hormone deficiency may also be called central hyphothyroidism.
Sometimes the symptoms of thyroid-stimulating hormone deficiency are not noticed. Low thyroid hormone levels may cause slow growth and delayed puberty, as well as other symptoms. Learn more about the symptoms of hypothyroidism.
Luteinizing hormone or follicle-stimulating hormone imbalance: Luteinizing hormone and follicle-stimulating hormone control reproduction. Abnormal levels of these hormones can cause different health problems. The type of problem depends on the radiation dose and whether hormone levels are higher or lower than normal.
Childhood cancer survivors who had brain tumors near the hypothalamus or pituitary gland, who were treated with lower doses of radiation to the brain, or who have hydrocephalus may develop central precocious puberty (a condition that causes puberty to start before age 8 in girls and 9 in boys). Central precocious puberty happens when the brain releases luteinizing and follicle-stimulating hormones too soon in the child’s development. This condition may be treated with gonadotropin-releasing hormone (GnRH) agonist therapy to delay puberty and help the child’s growth.
Childhood cancer survivors who were treated with higher doses of radiation to the brain may have low levels of luteinizing hormone or follicle-stimulating hormone. This condition may be treated with sex hormone replacement therapy. The dose will depend on the child’s age and whether the child has reached puberty.
Central diabetes insipidus: Central diabetes insipidus may be caused by the absence of or low amounts of all of the hormones made in the front part of the pituitary gland and released into the blood. It may occur in childhood cancer survivors treated with surgery in the area of the hypothalamus or pituitary gland. Symptoms of central diabetes insipidus may include:
Symptoms of central diabetes insipidus in infants may include:
vomiting
diarrhea
irritability
slowed growth and development
weight loss for no known reason
Certain tests and procedures are used to diagnose health problems in the neuroendocrine system.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Blood chemistry study uses a blood sample to measure the amounts of certain substances, such as glucose, released into the blood by organs and tissues in the body. An unusual amount of a substance can be a sign of disease.
Blood hormone studies use a blood sample to measure the amounts of certain hormones released into the blood by organs and tissues in the body. An unusual amount of a substance can be a sign of disease in the organ or tissue that makes it. The blood may be checked for abnormal levels of follicle-stimulating hormone, luteinizing hormone, estradiol, testosterone, cortisol, or free thyroxine (T4).
Lipid profile studies use a blood sample to measure the amounts of triglycerides, cholesterol, and low- and high-density lipoprotein cholesterol in the blood.
Talk to the doctor about whether tests and procedures are needed to check for signs of neuroendocrine late effects. If tests are needed, find out how often they should be done.
Metabolic syndrome
Metabolic syndrome is more likely to occur after treatment for certain childhood cancers.
Metabolic syndrome is a group of medical conditions that includes having too much fat around the abdomen and at least two of the following:
high levels of triglycerides and low levels of high-density lipoprotein (HDL) cholesterol in the blood
high levels of glucose (sugar) in the blood
Treatment for these and other childhood cancers may cause metabolic syndrome to occur later in life:
acute lymphoblastic leukemia (ALL)
cancers treated with a stem cell transplant
cancers treated with radiation to the abdomen, such as Wilms tumor or neuroblastoma
Radiation therapy increases the risk of metabolic syndrome.
The risk of metabolic syndrome may be increased in childhood cancer survivors after treatment with:
radiation therapy to the brain, abdomen, or pelvis
total-body irradiation (TBI) as part of a stem cell transplant
chemotherapy, such as alkylating agents
older age
Certain tests and procedures are used to diagnose metabolic syndrome.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Blood chemistry studies use a blood sample to measure the amounts of certain substances, such as glucose, released into the blood by organs and tissues in the body. An unusual amount of a substance can be a sign of disease.
Lipid profile studies use a blood sample to measure the amounts of triglycerides, cholesterol, and low- and high-density lipoprotein cholesterol in the blood.
Talk to the doctor about whether tests and procedures are needed to check for signs of metabolic syndrome. If tests are needed, find out how often they should be done.
Metabolic syndrome may cause heart and blood vessel disease and diabetes.
Weight changes and frailty are late effects that are more likely to occur after treatment for certain childhood cancers.
Weight changes include being underweight or having excess body weight or obesity. Frailty is a condition that usually affects older adults and includes having fatigue, slow walking speed, and muscle weakness.
Treatment for these and other childhood cancers may cause a change in weight or frailty:
Childhood cancer survivors who get enough exercise and have a normal amount of anxiety have a lower risk of obesity.
The risk of frailty increases after treatment with:
radiation therapy to the brain
TBI
Certain tests and procedures are used to diagnose a change in weight.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Blood chemistry studies use a blood sample to measure the amounts of certain substances, such as glucose, released into the blood by organs and tissues in the body. An unusual amount of a substance can be a sign of disease.
Lipid profile studies use a blood sample to measure the amounts of triglycerides, cholesterol, and low- and high-density lipoprotein cholesterol in the blood.
Being underweight, having excess body weight, or having obesity may be measured by weight, body mass index, percent of body fat, or size of the abdomen (belly fat).
Talk to the doctor about whether tests and procedures are needed to check for signs of a change in weight. If tests are needed, find out how often they should be done.
Immune System
Key Points
Cancer treatments and surgery to remove the spleen increase the risk of immune system late effects.
Late effects that affect the immune system may cause infection.
Children who have had their spleen removed or have lost vaccine immunity may need antibiotics or booster vaccine doses to lessen the risk of infection.
Cancer treatments and surgery to remove the spleen increase the risk of immune system late effects.
The risk of health problems that affect the immune system increases after treatment that affects the spleen, including:
stem cell transplant with or without graft-versus-host disease
Late effects that affect the immune system may cause infection.
Late effects that affect the immune system may increase the risk of viral infections or very serious bacterial infections. This risk is higher in younger children than in older children and in children who have decreased vaccine coverage after cancer treatment. The risk may be greater in the early years after the spleen stops working or is removed by surgery. These symptoms may be caused by infection:
redness, swelling, or warmth of a part of the body
pain that is in one part of the body, such as the eye, ear, or throat
fever
An infection may cause other symptoms that depend on the part of the body affected. For example, a lung infection may cause a cough and trouble breathing.
Children who have had their spleen removed or have lost vaccine immunity may need antibiotics or booster vaccine doses to lessen the risk of infection.
Daily antibiotics may be prescribed for children younger than 5 years whose spleen is no longer working or for at least 1 year after surgery to remove the spleen. For certain high-risk patients, daily antibiotics may be prescribed throughout childhood and into adulthood.
In addition, children with an increased risk of infection should be vaccinated on a schedule through adolescence against:
Some children may lose the protection they had from vaccines during their cancer treatments. Talk to the doctor about whether childhood vaccinations given before cancer treatment need to be repeated.
Musculoskeletal System
Key Points
Bone and joint late effects are more likely to occur after treatment for certain childhood cancers.
Radiation therapy, surgery, chemotherapy, and other treatments increase the risk of bone and joint late effects.
Radiation therapy
Surgery
Chemotherapy and other drug therapy
Stem cell transplant
Possible symptoms of bone and joint late effects include swelling over a bone or bone and joint pain.
Certain tests and procedures are used to diagnose health problems in the bone and joint.
Bone and joint late effects are more likely to occur after treatment for certain childhood cancers.
Treatment for these and other childhood cancers may cause bone and joint late effects:
Poor nutrition and not enough exercise may also cause bone late effects.
Radiation therapy, surgery, chemotherapy, and other treatments increase the risk of bone and joint late effects.
Radiation therapy
Radiation therapy can stop or slow the growth of bone and muscle. The type of bone and joint late effects depends on the part of the body that received radiation therapy. Radiation therapy may cause:
changes in the way the face or skull form, especially when high-dose radiation with or without chemotherapy is given to children before age 5 years
Amputation or limb-sparing surgery to remove the cancer and prevent it from coming back may cause late effects depending on where the tumor was, age of the patient, and type of surgery. Health problems after amputation or limb-sparing surgery may include:
weak or thin bones that can break easily (osteoporosis)
one or more parts of a bone to die from a lack of blood flow (osteonecrosis), especially in the hip or knee
Stem cell transplant
A stem cell transplant can affect the bone and joints in different ways:
Total-body irradiation given as part of a stem cell transplant may affect the body’s ability to make growth hormone and cause short stature (being shorter than normal). It may also cause weak or thin bones that can break easily (osteoporosis) and the formation of a benign tumor called osteochondroma.
Chronicgraft-versus-host disease that may occur after a stem cell transplant can cause the muscles around joints to tighten, leading to stiff joints (joint contractures). Chronic graft-versus-host disease may also cause one or more parts of a bone to die from a lack of blood flow (osteonecrosis).
Possible symptoms of bone and joint late effects include swelling over a bone or bone and joint pain.
These and other symptoms may be caused by bone and joint late effects or by other problems:
swelling over a bone or bony part of the body
pain in a bone or joint
redness or warmth over a bone or joint
joint stiffness or trouble moving normally
a bone that breaks easily or for no known reason
short stature (being shorter than normal)
one side of the body looks higher than the other side or the body tilts to one side
always sitting or standing in a slouching position or having the appearance of a hunched back
The only way to know if these symptoms are caused by bone and joint late effects is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the bone and joint.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Bone mineral density scan is an imaging test that measures bone density (the amount of bone mineral in a certain amount of bone) by passing x-rays with two different energy levels through the bone. It is used to diagnose osteoporosis (weak or thin bones that can break easily). Also called BMD scan, DEXA, DEXA scan, dual energy x-ray absorptiometric scan, dual x-ray absorptiometry, and DXA.
X-ray is a type of radiation that can go through the body and make pictures of areas inside the body, such as bones.
Vitamin D test uses a sample of blood to measure the amount of vitamin D in the body. Vitamin D is a nutrient the body needs to build and maintain strong bones.
Calcium blood test uses a sample of blood to measure the amount of calcium in the body. Calcium is a mineral the body needs to build and maintain strong bones.
Talk to the doctor about whether tests and procedures are needed to check for signs of bone and joint late effects. If tests are needed, find out how often they should be done.
Reproductive System
Key Points
Reproductive system late effects are more likely to occur after treatment for certain childhood cancers.
Surgery, radiation therapy, and certain types of chemotherapy increase the risk of late effects that affect the reproductive system.
Testicles
Late effects that affect the testicles may cause certain health problems.
Certain tests and procedures are used to diagnose health problems in the testicles.
Ovaries
Late effects that affect the ovaries may cause certain health problems.
Possible symptoms of ovarian late effects include irregular or absent menstrual periods and hot flashes.
Certain tests and procedures are used to diagnose health problems in the ovaries.
Fertility and reproduction
Childhood cancer survivors may have late effects that affect pregnancy.
There are methods that may be used to help childhood cancer survivors have children.
Children of childhood cancer survivors are not affected by the parent’s previous treatment for cancer.
Reproductive system late effects are more likely to occur after treatment for certain childhood cancers.
Treatment for these and other childhood cancers may cause testicular late effects:
total-body irradiation, high-dose chemotherapy, or both before a stem cell transplant
radiation therapy to the abdomen or pelvis with alkylating agents
In female survivors who had radiation therapy to the abdomen, the damage to the ovaries depends on the radiation dose, age at the time of treatment, and whether all or part of the abdomen received radiation.
Testicles
Late effects that affect the testicles may cause certain health problems.
Late effects of the testicles and related health problems include:
Low sperm count: A zero sperm count or a low sperm count may be temporary or permanent. This depends on the radiation dose and schedule, the area of the body treated, and the age when treated.
Low testosterone levels: This can lead to underdeveloped testicles, infertility, and sexual problems in adulthood.
After treatment with chemotherapy or radiation, the body’s ability to make sperm may come back over time.
Certain tests and procedures are used to diagnose health problems in the testicles.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests:
Blood hormone studies use a blood sample to measure the amounts of certain hormones released into the blood by organs and tissues in the body. An unusual amount of a substance can be a sign of disease in the organ or tissue that makes it. The blood may be checked for abnormal levels of follicle-stimulating hormone, luteinizing hormone, testosterone.
Semen analysis (sperm count) measures the amount and quality of semen and sperm to see if there are problems that are causing infertility.
Ovaries
Late effects that affect the ovaries may cause certain health problems.
Ovarian late effects and other health-related problems include:
early menopause, especially in women who had their ovaries removed or were treated with both an alkylating agent and radiation therapy to the abdomen
sex traits, such as developing arm, pubic, and leg hair or having the breasts enlarge, do not occur at puberty
weak or thin bones that can break easily (osteoporosis)
inability to get pregnant
The only way to know if these symptoms are caused by late effects to the ovaries is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the ovaries.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following test.
Blood hormone studies use a blood sample to measure the amounts of certain hormones released into the blood by organs and tissues in the body. An unusual amount of a substance can be a sign of disease in the organ or tissue that makes it. The blood may be checked for abnormal levels of follicle-stimulating hormone, luteinizing hormone, estradiol, or TSH.
Fertility and reproduction
Childhood cancer survivors may have late effects that affect pregnancy.
Late effects on pregnancy include increased risk of:
the fetus is not in the right position for birth (for example, the foot or buttock is in position to come out before the head)
peripartum cardiomyopathy
Long-term survivors of childhood, adolescent, and young adult cancers who have received radiation therapy or certain types of chemotherapy are at risk of a rare type of heart failure during pregnancy called peripartum cardiomyopathy. It may occur during late pregnancy and up to 5 months after giving birth. If cancer treatments have caused damage to the heart, these patients may also be at risk of congestive heart failure during pregnancy. Patients at risk of these conditions should be closely followed for weakness of the heart muscle during pregnancy.
Some studies have not shown an increased risk of late effects on pregnancy.
There are methods that may be used to help childhood cancer survivors have children.
The following methods may be used so that childhood cancer survivors can have children:
freezing the eggs or sperm before cancer treatment in patients who have reached puberty (egg and sperm banking)
preventing damage to the ovaries before radiation therapy by moving one or both ovaries and fallopian tubes out of the field of radiation and attaching them to the wall of the abdomen (ovarian transposition)
fertilizing an egg outside the body by injecting a sperm into it (intracytoplasmic sperm injection)
in vitro fertilization (IVF), which is a procedure where eggs and sperm are placed together in a container, giving the sperm the chance to enter an egg
Children of childhood cancer survivors are not affected by the parent’s previous treatment for cancer.
The children of childhood cancer survivors do not appear to have an increased risk of birth defects, genetic disease, or cancer.
Respiratory System
Key Points
Lung late effects are more likely to occur after treatment for certain childhood cancers.
Certain types of chemotherapy and radiation to the lungs increase the risk of lung late effects.
Late effects that affect the lungs may cause certain health problems.
Possible symptoms of lung late effects include trouble breathing and cough.
Certain tests and procedures are used to diagnose health problems in the lung.
Health habits that promote healthy lungs are important for survivors of childhood cancer.
Lung late effects are more likely to occur after treatment for certain childhood cancers.
Treatment for these and other childhood cancers may cause lung late effects:
In survivors who had radiation to the chest, the damage to the lungs and chest wall depends on the radiation dose, whether all or part of the lungs and chest wall received radiation, whether the radiation was given in small, divided daily doses, and the child’s age at treatment.
The risk of lung late effects is greater in childhood cancer survivors who are treated with a combination of surgery, chemotherapy, and/or radiation therapy. The risk is also increased in survivors who have a history of:
congestion (a feeling of fullness in the lungs from extra mucus)
chronic lung infections
feeling tired
The only way to know if these symptoms are caused by lung late effects is to see a doctor.
Lung late effects in childhood cancer survivors may occur slowly over time or there may be no symptoms. Sometimes lung damage can be detected only by imaging or pulmonary function testing. Lung late effects may improve over time.
Certain tests and procedures are used to diagnose health problems in the lung.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Chest x-ray is an x-ray of the organs and bones inside the chest. An x-ray is a type of radiation that can go through the body and make pictures of areas inside the body.
Pulmonary function test (PFT) is a test to see how well the lungs are working. It measures how much air the lungs can hold and how quickly air moves into and out of the lungs. It also measures how much oxygen is used and how much carbon dioxide is given off during breathing. This is also called lung function test.
CT scan (CAT scan) uses a computer linked to an x-ray machine to make a series of detailed pictures of areas inside the body, such as the chest. The pictures are taken from different angles and are used to create 3-D views of tissues and organs. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography. Learn more about Computed Tomography (CT) Scans and Cancer.
Talk to the doctor about whether tests and procedures are needed to check for signs of lung late effects. If tests are needed, find out how often they should be done.
Health habits that promote healthy lungs are important for survivors of childhood cancer.
Childhood cancer survivors with lung late effects should take care to protect their health, including:
The risk of hearing loss from radiation therapy is greater in children who are diagnosed with cancer before age 5 years, and the risk increases for children of all ages with higher radiation doses. The risk of hearing loss is also greater in childhood cancer survivors who received radiation therapy to the brain and chemotherapy at the same time.
The risk of hearing loss may be lower if children treated with cisplatin were also given a drug called sodium thiosulfate.
Hearing loss is the most common sign of hearing late effects.
These and other symptoms may be caused by hearing late effects or by other problems:
hearing loss
ringing in the ears
feeling dizzy
too much hardened wax in the ear
The only way to know if these symptoms are caused by hearing late effects is to see a doctor.
Hearing loss may occur during treatment, soon after treatment ends, or several months or years after treatment ends and worsen over time. A young child may have trouble learning, communicating, doing well in school, and interacting with others if they have hearing loss. In adulthood, hearing loss can lead to social isolation, depression, anxiety, and trouble finding stable employment. Talk to a doctor to learn about hearing aids or other ways to cope with hearing loss.
Certain tests and procedures are used to diagnose health problems in the ear and hearing problems.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Otoscopic exam is an exam of the ear. An otoscope is used to look at the ear canal and the eardrum to check for signs of infection or hearing loss. Sometimes the otoscope has a plastic bulb that is squeezed to release a small puff of air into the ear canal. In a healthy ear, the eardrum will move. If there is fluid behind the eardrum, it will not move.
Hearing test can be done in different ways depending on a person’s age. The test is done to check if the person can hear soft and loud sounds and low- and high-pitched sounds. Each ear is checked separately.
Talk to the doctor about whether tests and procedures are needed to check for signs of hearing late effects. If tests are needed, find out how often they should be done.
Seeing
Eye and vision problems are a late effect that is more likely to occur after treatment for certain childhood cancers.
Treatment for these and other childhood cancers may cause eye and vision late effects:
Possible signs and symptoms of eye and vision late effects include changes in vision and dry eyes.
These and other symptoms may be caused by eye and vision late effects or by other problems:
changes in vision, such as:
not being able to see objects that are close
not being able to see objects that are far away
double vision
cloudy or blurred vision
colors seem faded
being sensitive to light or trouble seeing at night
seeing a glare or halo around lights at night
dry eyes that may feel like they are itchy, burning, or swollen, or like there is something in the eye
eye pain
eye redness
having a growth on the eyelid
drooping of the upper eyelid
The only way to know if these symptoms are caused by eye and vision late effects is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the eye and vision problems.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Eye exam with dilated pupil is when the pupil is dilated (widened) with medicated eye drops to allow the doctor to look through the lens and pupil to the retina. The inside of the eye, including the retina and the optic nerve, is checked using an instrument that makes a narrow beam of light. This is sometimes called a slit-lamp exam. If there is a tumor, the doctor may take pictures over time to keep track of changes in the size of the tumor and how fast it is growing.
Indirect ophthalmoscopy is an exam of the inside of the back of the eye using a small magnifying lens and a light.
Talk to the doctor about whether tests and procedures are needed to check for signs of eye and vision late effects. If tests are needed, find out how often they should be done.
Urinary System
Key Points
Kidney
Kidney problems are a late effect that is more likely to occur after treatment for certain childhood cancers.
Certain types of chemotherapy increase the risk of kidney late effects.
Late effects that affect the kidney may cause certain health problems.
Possible symptoms of kidney late effects include problems urinating and swelling of the feet or hands.
Certain tests and procedures are used to diagnose health problems in the kidney.
Health habits that promote healthy kidneys are important for survivors of childhood cancer.
Bladder
Bladder problems are a late effect that is more likely to occur after treatment for certain childhood cancers.
Surgery to the pelvic area and certain types of chemotherapy increase the risk of bladder late effects.
Late effects that affect the bladder may cause certain health problems.
Possible symptoms of bladder late effects include changes in urination and swelling of the feet or hands.
Certain tests and procedures are used to diagnose health problems in the bladder.
Kidney
Kidney problems are a late effect that is more likely to occur after treatment for certain childhood cancers.
Treatment for these and other childhood cancers may cause kidney late effects:
The risk of kidney late effects is greater in childhood cancer survivors who were treated with a combination of surgery, chemotherapy, or radiation therapy.
The following may also increase the risk of kidney late effects:
Sometimes there are no signs or symptoms in the early stages. Signs or symptoms may appear as damage to the kidney continues over time. The only way to know if these symptoms are caused by kidney late effects is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the kidney.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Blood chemistry study uses a blood sample to measure the amounts of certain substances, such as creatinine, released into the blood by organs and tissues in the body. An unusual amount of a substance may be a sign of kidney disease.
Ultrasound exam uses high-energy sound waves (ultrasound) that bounce off internal tissues or organs, such as the kidney, and make echoes. The echoes form a picture of body tissues called a sonogram.
Talk to the doctor about whether tests and procedures are needed to check for signs of kidney late effects. If tests are needed, find out how often they should be done.
If kidney problems are found, treatment options are available, including kidney transplant.
Health habits that promote healthy kidneys are important for survivors of childhood cancer.
Childhood cancer survivors who had all or part of their kidney removed should talk to their doctor about:
whether it is safe to play sports that have a high risk of heavy contact or impact such as football or hockey
bicycle safety and avoiding handlebar injuries
wearing a seatbelt around the hips, not the waist
Bladder
Bladder problems are a late effect that is more likely to occur after treatment for certain childhood cancers.
Treatment for these and other childhood cancers may cause bladder late effects:
a blockage in the kidney, ureter, bladder, or urethra
urinary tract infections that last a long time or keep coming back
Possible symptoms of bladder late effects include changes in urination and swelling of the feet or hands.
These and other symptoms may be caused by bladder late effects or by other problems:
feeling the need to urinate without being able to do so
frequent urination (especially at night)
trouble urinating
feeling like the bladder does not empty completely after urination
swelling of the legs, ankles, feet, face, or hands
little or no bladder control
blood in the urine
The only way to know if these symptoms are caused by bladder late effects is to see a doctor.
Certain tests and procedures are used to diagnose health problems in the bladder.
In addition to asking the childhood cancer survivor about their personal and family health history and doing a physical exam, the doctor may perform the following tests and procedures:
Blood chemistry study uses a blood sample to measure the amounts of certain substances, such as magnesium, calcium, and potassium, released into the blood by organs and tissues in the body. An unusual amount of a substance may be a sign of bladder problems.
Urinalysis checks the color of urine and its contents, such as sugar, protein, red blood cells, and white blood cells.
Urine culture checks for bacteria, yeast, or other microorganisms in the urine when there are symptoms of infection. Urine cultures can help identify the type of microorganism that is causing an infection. Treatment of the infection depends on the type of microorganism that is causing the infection.
Ultrasound exam uses high-energy sound waves (ultrasound) that bounce off internal tissues or organs, such as the bladder, and make echoes. The echoes form a picture of body tissues called a sonogram.
Talk to the doctor about whether tests and procedures are needed to check for signs of bladder late effects. If tests are needed, find out how often they should be done.
To Learn More About Late Effects of Treatment for Childhood Cancer
For more information about late effects of treatment for childhood cancer:
Physician Data Query (PDQ) is the National Cancer Institute’s (NCI’s) comprehensive cancer information database. The PDQ database contains summaries of the latest published information on cancer prevention, detection, genetics, treatment, supportive care, and complementary and alternative medicine. Most summaries come in two versions. The health professional versions have detailed information written in technical language. The patient versions are written in easy-to-understand, nontechnical language. Both versions have cancer information that is accurate and up to date and most versions are also available in Spanish.
PDQ is a service of the NCI. The NCI is part of the National Institutes of Health (NIH). NIH is the federal government’s center of biomedical research. The PDQ summaries are based on an independent review of the medical literature. They are not policy statements of the NCI or the NIH.
Purpose of This Summary
This PDQ cancer information summary has current information about the late effects of treatment for childhood cancer. It is meant to inform and help patients, families, and caregivers. It does not give formal guidelines or recommendations for making decisions about health care.
Reviewers and Updates
Editorial Boards write the PDQ cancer information summaries and keep them up to date. These Boards are made up of experts in cancer treatment and other specialties related to cancer. The summaries are reviewed regularly and changes are made when there is new information. The date on each summary (“Updated”) is the date of the most recent change.
The information in this patient summary was taken from the health professional version, which is reviewed regularly and updated as needed, by the PDQ Pediatric Treatment Editorial Board.
Clinical Trial Information
A clinical trial is a study to answer a scientific question, such as whether one treatment is better than another. Trials are based on past studies and what has been learned in the laboratory. Each trial answers certain scientific questions in order to find new and better ways to help cancer patients. During treatment clinical trials, information is collected about the effects of a new treatment and how well it works. If a clinical trial shows that a new treatment is better than one currently being used, the new treatment may become “standard.” Patients may want to think about taking part in a clinical trial. Some clinical trials are open only to patients who have not started treatment.
Clinical trials can be found online at NCI’s website. For more information, call the Cancer Information Service (CIS), NCI’s contact center, at 1-800-4-CANCER (1-800-422-6237).
Permission to Use This Summary
PDQ is a registered trademark. The content of PDQ documents can be used freely as text. It cannot be identified as an NCI PDQ cancer information summary unless the whole summary is shown and it is updated regularly. However, a user would be allowed to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks in the following way: [include excerpt from the summary].”
The best way to cite this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Late Effects of Treatment for Childhood Cancer. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/childhood-cancers/late-effects-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389365]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use in the PDQ summaries only. If you want to use an image from a PDQ summary and you are not using the whole summary, you must get permission from the owner. It cannot be given by the National Cancer Institute. Information about using the images in this summary, along with many other images related to cancer can be found in Visuals Online. Visuals Online is a collection of more than 3,000 scientific images.
Disclaimer
The information in these summaries should not be used to make decisions about insurance reimbursement. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s E-mail Us.
Targeted therapy treats cancer by targeting proteins that control how cancer cells grow, divide, and spread.
Credit: National Cancer Institute
What is targeted therapy?
Targeted therapy is a type of cancer treatment that targets proteins that control how cancer cells grow, divide, and spread. It is the foundation of precision medicine. As researchers learn more about the DNA changes and proteins that drive cancer, they are better able to design treatments that target these proteins.
Small-molecule drugs are small enough to enter cells easily, so they are used for targets that are inside cells.
Monoclonal antibodies, also known as therapeutic antibodies, are proteins produced in the lab. These proteins are designed to attach to specific targets found on cancer cells. Some monoclonal antibodies mark cancer cells so that they will be better seen and destroyed by the immune system. Other monoclonal antibodies directly stop cancer cells from growing or cause them to self-destruct. Still others carry toxins to cancer cells. Learn more about monoclonal antibodies.
How Monoclonal Antibodies Treat Cancer
Learn how monoclonal antibodies such as trastuzumab, pembrolizumab, and rituximab are used to treat cancer.
Who is treated with targeted therapy?
For some types of cancer, such as chronic myelogenous leukemia (also known as CML), most people with that cancer will have a target for a certain drug, so they can be treated with that drug. But most of the time, your tumor will need to be tested to see if it contains targets for which there is a drug.
Testing your cancer for targets that could help choose your treatment is called biomarker testing. See Biomarker Testing for Cancer Treatment for more information.
You may need to have a biopsy for biomarker testing. A biopsy is a procedure in which your doctor removes a piece of the tumor for testing. There are some risks to having a biopsy. These risks vary depending on the size of the tumor and where it is located. Your doctor will explain the risks of having a biopsy for your type of tumor.
Most types of targeted therapy help treat cancer by interfering with specific proteins that help tumors grow and spread throughout the body. This is different from chemotherapy, which often kills all cells that grow and divide quickly. The following explains the different ways that targeted therapy treats cancer.
Help the immune system destroy cancer cells. One reason that cancer cells thrive is because they can hide from your immune system. Certain targeted therapies can mark cancer cells so it is easier for the immune system to find and destroy them. Other targeted therapies help boost your immune system to work better against cancer. Learn more about immunotherapy to treat cancer.
Stop cancer cells from growing by interrupting signals that cause them to grow and divide without order. Healthy cells in your body usually divide to make new cells only when they receive strong signals to do so. These signals bind to proteins on the cell surface, telling the cells to divide. This process helps new cells form only as your body needs them. But, some cancer cells have changes in the proteins on their surface that tell them to divide whether or not signals are present. Some targeted therapies interfere with these proteins, preventing them from telling the cells to divide. This process helps slow cancer’s uncontrolled growth.
Stop signals that help form blood vessels. To grow beyond a certain size, tumors need to form new blood vessels in a process called angiogenesis. The tumor sends signals that start angiogenesis. Some targeted therapies called angiogenesis inhibitors interfere with these signals to prevent a blood supply from forming. Without a blood supply, tumors stay small. Or, if a tumor already has a blood supply, these treatments can cause blood vessels to die, which causes the tumor to shrink. Learn more about angiogenesis inhibitors.
Deliver cell-killing substances to cancer cells. Some monoclonal antibodies are combined with cell-killing substances such as toxins, chemotherapy drugs, or radiation. Once these monoclonal antibodies attach to targets on the surface of cancer cells, the cells take up the cell-killing substances, causing them to die. Cells that don’t have the target will not be harmed.
Cause cancer cell death. Healthy cells die in an orderly manner when they become damaged or are no longer needed. But, cancer cells have ways of avoiding this dying process. Some targeted therapies can cause cancer cells to go through this process of cell death, which is called apoptosis.
Starve cancer of hormones it needs to grow. Some breast and prostate cancers require certain hormones to grow. Hormone therapies are a type of targeted therapy that can work in two ways. Some hormone therapies prevent your body from making specific hormones. Others prevent the hormones from acting on your cells, including cancer cells. Learn more about hormone therapy for prostate cancer and hormone therapy for breast cancer.
Are there drawbacks to targeted therapy?
Targeted therapy does have some drawbacks.
Cancer cells can become resistant to targeted therapy. Resistance can happen when the target itself changes and the targeted therapy is not able to interact with it. Or it can happen when cancer cells find new ways to grow that do not depend on the target. Because of resistance, targeted therapy may work best when used with more than one type of targeted therapy or with other cancer treatments, such as chemotherapy and radiation.
Drugs for some targets are hard to develop. Reasons include the target’s structure, the target’s function in the cell, or both.
What are the side effects of targeted therapy?
When targeted therapy was first developed, scientists thought that it would be less toxic than chemotherapy. But they have learned that targeted therapy can also cause serious side effects. The side effects that you may have depends on the type of targeted therapy you receive and how your body reacts to it.
The most common side effects of targeted therapy include diarrhea and liver problems. Other side effects might include
Small-molecule drugs are pills or capsules that you can swallow.
Monoclonal antibodies are usually given through a needle in a blood vein.
Where do I go for targeted therapy?
Where you go for treatment depends on which drugs you are getting and how they are given. You may take targeted therapy at home. Or you may receive targeted therapy in a doctor’s office, clinic, or outpatient unit in a hospital. Outpatient means you do not spend the night in the hospital.
How often will I receive targeted therapy?
How often and how long you receive targeted therapy depends on
your type of cancer and how advanced it is
the type of targeted therapy
how your body reacts to treatment
You may have treatment every day, every week, or every month. Some targeted therapies are given in cycles. A cycle is a period of treatment followed by a period of rest. The rest period gives your body a chance to recover and build new healthy cells.
How will targeted therapy affect me?
Targeted therapy affects people in different ways. How you feel depends on how healthy you are before treatment, your type of cancer, how advanced it is, the kind of targeted therapy you are getting, and the dose. Doctors and nurses cannot know for certain how you will feel during treatment.
How will I know whether targeted therapy is working?
While you are receiving targeted therapy, you will see your doctor often. He or she will give you physical exams and ask you how you feel. You will have medical tests, such as blood tests, x-rays, and different types of scans. These regular visits and tests will help the doctor know whether the treatment is working.
Where can I find out about clinical trials of targeted therapy?
Clinical trials of targeted therapy and other cancer treatments take place in cities and towns across the United States and throughout the world. They take place in doctors’ offices, cancer centers, medical centers, community hospitals and clinics, and veteran and military hospitals.
To find clinical trials of targeted therapy use this advanced search form. Under “Keywords/Phrases,” type “targeted therapy.” Under “Trial Type,” select the box for “Treatment” trials.
Having cancer brings many changes to a child’s life. You can help your child by keeping her life as normal as possible.
Credit: iStock
A cancer diagnosis is upsetting at any age, but especially so when the patient is a child. It’s natural to have many questions, such as, Who should treat my child? Will my child get well? What does all of this mean for our family? Not all questions have answers, but the information and resources on this page provide a starting point for understanding the basics of childhood cancer.
Types of Cancer in Children
In the United States in 2025, an estimated 9,550 new cases of cancer will be diagnosed among children from birth to 14 years, and about 1,050 children are expected to die from the disease. Although cancer death rates for this age group have declined by 70 percent from 1970 through 2020, cancer remains the leading cause of death from disease among children. The most common types of cancer diagnosed in children ages 0 to 14 years are leukemias, brain and other central nervous system (CNS) tumors, and lymphomas.
NCI’s Cancer Stat Facts include detailed cancer rate and trend information for certain types of childhood cancer.
Treating Childhood Cancer
Children’s cancers are not always treated like adult cancers. Pediatric oncology is a medical specialty focused on the care of children with cancer. It’s important to know that this expertise exists and that there are effective treatments for many childhood cancers.
Treatment Considerations for Children with Cancer
Pediatric oncology experts and parents discuss childhood cancer treatment-related decisions, side effects, clinical trials for children with cancer, and strategies to care for children at home.
There are many types of cancer treatment. The types of treatment that a child with cancer receives will depend on the type of cancer and how advanced it is. Common treatments include: surgery, chemotherapy, radiation therapy, immunotherapy, and stem cell transplant. Learn about these and other therapies in our Types of Cancer Treatment section.
Our summary about Childhood Cancer Genomics describes the genomic alterations associated with different pediatric cancers, and their significance for therapy and prognosis.
Before any new treatment can be made widely available to patients, it must be studied in clinical trials (research studies) and found to be safe and effective in treating disease. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials.
Children face unique issues during their treatment for cancer, after the completion of treatment, and as survivors of cancer. For example, they may receive more intense treatments, cancer and its treatments have different effects on growing bodies than adult bodies, and they may respond differently to drugs that control symptoms in adults. Late effects of treatment are discussed later on this page in the Survivorship section.
Where Children with Cancer Are Treated
Children who have cancer are often treated at a children’s cancer center, which is a hospital or unit in a hospital that specializes in treating children with cancer.
The doctors and other health professionals at these centers have special training and expertise to give complete care to children. Specialists at a children’s cancer center are likely to include primary care physicians, pediatric oncologists/hematologists, pediatric surgical specialists, radiation oncologists, rehabilitation specialists, pediatric nurse specialists, social workers, and psychologists. At these centers, clinical trials are available for most types of cancer that occur in children, and the opportunity to participate in a trial is offered to many patients.
Hospitals that have experts in treating children with cancer are usually member institutions of the NCI-supported Children’s Oncology Group (COG). COG is the world’s largest organization that conducts clinical research to improve the care and treatment of children with cancer. NCI’s Cancer Information Service can help families find COG-affiliated hospitals.
At the NIH Clinical Center in Bethesda, Maryland, NCI’s Pediatric Oncology Branch cares for children and young adults with cancer. Health professionals and scientists conduct translational research that spans basic science to clinical trials to improve outcomes for children and young adults with cancer and genetic tumor predisposition syndromes.
Coping with Cancer
Adjusting to a child’s cancer diagnosis and finding ways to stay strong is challenging for everyone in a family. Our page, Support for Families When a Child Has Cancer, has tips for talking with children about their cancer and preparing them for changes they may experience. Also included are ways to help brothers and sisters cope, steps parents can take when they need support, and tips for working with the health care team. Various aspects of coping and support are also discussed in the publication Children with Cancer: A Guide for Parents.
Survivorship
It’s essential for childhood cancer survivors to receive follow-up care to monitor their health after completing treatment. All survivors should have a treatment summary and a survivorship care plan, as discussed on our Care for Childhood Cancer Survivors page. That page also has information on clinics that specialize in providing follow-up care for people who have had childhood cancer.
Survivors of any kind of cancer can develop health problems months or years after cancer treatment, known as late effects, but late effects are of particular concern for childhood cancer survivors because treatment of children can lead to profound, lasting physical and emotional effects. Late effects vary with the type of cancer, the child’s age, the type of treatment, and other factors. Information on types of late effects and ways to manage these can be found on our Care for Childhood Cancer Survivors page. The PDQ® Late Effects of Treatment for Childhood Cancer summary has in-depth information.
The causes of most childhood cancers are not known. About 8 to 10 percent of all cancers in children are caused by an inherited mutation (a genetic mutation that can be passed from parents to their children).
Most cancers in children, like those in adults, are thought to develop as a result of mutations in genes that lead to uncontrolled cell growth and eventually cancer. In adults, these gene mutations reflect the cumulative effects of aging and long-term exposure to cancer-causing substances. However, identifying potential environmental causes of childhood cancer has been difficult, partly because cancer in children is rare and partly because it is difficult to determine what children might have been exposed to early in their development. More information about possible causes of cancer in children is available in the fact sheet, Cancer in Children and Adolescents.
Research
NCI supports a broad range of research to better understand the causes, biology, and patterns of childhood cancers and to identify the best ways to successfully treat children with cancer. In the context of clinical trials, researchers are treating and learning from young cancer patients. Researchers are also following childhood cancer survivors to learn about health and other issues they may face as a result of their cancer treatment. To learn more, see Research on Childhood Cancers.
Carcinoma of unknown primary is a disease in which malignant (cancer) cells are found in the body but the place the cancer began is not known.
The signs and symptoms of disease are caused by the metastatic cancer and depend on where the cancer has spread.
Blood and imaging tests are done to learn more about the cause of the signs and symptoms.
A biopsy is done to diagnose metastatic cancer.
Because the place where the cancer started is not known, more tests and procedures are done to search for the primary cancer.
When tests are able to find the primary cancer, the cancer is no longer a CUP and treatment is based on the type of primary cancer.
Sometimes the primary cancer is never found.
Carcinoma of unknown primary is a disease in which malignant (cancer) cells are found in the body but the place the cancer began is not known.
Cancer can form in any tissue in the body. The first cancer to form is called the primary cancer. The process of cancer cells spreading to other parts of the body is called metastasis. The cancer that has spread to another part of the body is called metastatic cancer.
EnlargeIn cancer of unknown primary, cancer cells have spread in the body but the place where the primary cancer began is not known.
Many cancer deaths are caused when cancer moves from the original tumor and spreads to other tissues and organs. This is called metastatic cancer. This animation shows how cancer cells travel from the place in the body where they first formed to other parts of the body.
The signs and symptoms of disease are caused by the metastatic cancer and depend on where the cancer has spread.
The primary cancer does not cause signs and symptoms of disease.
Check with your doctor if you have any of the following general signs of cancer:
Lump or thickening in any part of the body.
Pain that is in one part of the body and does not go away.
A cough that does not go away or hoarseness in the voice.
The pathologist views the tissue to look for cancer cells and to find out the type of cancer. Cancer cells usually look like the cells in the type of tissue in which the cancer began. In CUP, the cancer cells do not look like the cells of the tissue they were found in. The pathologist cannot determine the type of primary cancer.
One or more of the following laboratory tests may be used to further study the tissue samples:
Genetic analysis: A laboratory test in which the DNA in a sample of cancer cells or tissue is studied to check for mutations (changes) that may help predict the best treatment for CUP.
Histologic study: A laboratory test in which stains are added to a sample of cancer cells or tissue and viewed under a microscope to look for certain changes in the cells. Certain changes in the cells are linked to certain types of cancer.
Immunohistochemistry: A laboratory test that uses antibodies to check for certain antigens (markers) in a sample of a patient’s tissue. The antibodies are usually linked to an enzyme or a fluorescent dye. After the antibodies bind to a specific antigen in the tissue sample, the enzyme or dye is activated, and the antigen can then be seen under a microscope. This type of test is used to help diagnose cancer and to help tell one type of cancer from another type of cancer.
Reverse transcription–polymerase chain reaction (RT–PCR) test: A laboratory test in which the amount of a genetic substance called mRNA made by a specific gene is measured. An enzyme called reverse transcriptase is used to convert a specific piece of RNA into a matching piece of DNA, which can be amplified (made in large numbers) by another enzyme called DNA polymerase. The amplified DNA copies help tell whether a specific mRNA is being made by a gene. RT–PCR can be used to check the activation of certain genes that may indicate the presence of cancer cells. This test may be used to look for certain changes in a gene or chromosome, which may help diagnose cancer.
Cytogenetic analysis: A laboratory test in which the chromosomes of cells in a sample of tumor tissue are counted and checked for any changes, such as broken, missing, rearranged, or extra chromosomes. Changes in certain chromosomes may be a sign of cancer. Cytogenetic analysis is used to help diagnose cancer, plan treatment, or find out how well treatment is working. Changes in certain chromosomes are linked to certain types of cancer.
Light and electron microscopy: A laboratory test in which cells in a sample of tissue are viewed under regular and high-powered microscopes to look for certain changes in the cells.
Because the place where the cancer started is not known, more tests and procedures are done to search for the primary cancer.
The following tests and procedures may be done:
Physical exam and health history: An exam of the body to check general signs of health, including checking for signs of disease, such as lumps or anything else that seems unusual. A history of the patient’s health habits and past illnesses and treatments will also be taken.
CT scan (CAT scan): A procedure that makes a series of detailed pictures of areas inside the body, such as the chest or abdomen, taken from different angles. The pictures are made by a computer linked to an x-ray machine. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography.
MRI (magnetic resonance imaging): A procedure that uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the body. This procedure is also called nuclear magnetic resonance imaging (NMRI).
PET scan (positron emission tomography scan): A procedure to find malignant tumor cells in the body. A small amount of radioactive glucose (sugar) is injected into a vein. The PET scanner rotates around the body and makes a picture of where glucose is being used in the body. Malignant tumor cells show up brighter in the picture because they are more active and take up more glucose than normal cells do.
Ultrasound exam: A procedure in which high-energy sound waves (ultrasound) are bounced off internal tissues or organs and make echoes. The echoes form a picture of body tissues called a sonogram. The picture can be printed to be looked at later.
Endoscopy: A procedure to look at organs and tissues inside the body to check for abnormal areas. An endoscope is inserted through an incision (cut) in the skin or opening in the body, such as the mouth. An endoscope is a thin, tube-like instrument with a light and a lens for viewing. It may also have a tool to remove tissue or lymph node samples, which are checked under a microscope for signs of disease. For example, a colonoscopy may be done.
The primary cancer was removed during surgery for another condition and doctors didn’t know cancer had formed. For example, in adults, a uterus with cancer may be removed during a hysterectomy to treat a serious infection.
Because the primary cancer is unknown, it may be harder to choose the best treatment.
Stages of Childhood Carcinoma of Unknown Primary
The extent or spread of cancer is usually described as stages. The stage of the cancer is usually used to plan treatment. However, carcinoma of unknown primary (CUP) has already spread to other parts of the body when it is found. There is no standard staging system for CUP.
Sometimes childhood carcinoma of unknown primary recurs (comes back) after treatment.
Treatment Option Overview
Key Points
There are different types of treatment for children with carcinoma of unknown primary.
Children with carcinoma of unknown primary should have their treatment planned by a team of doctors who are experts in treating childhood cancer.
Three types of standard treatment are used:
Radiation therapy
Chemotherapy
Targeted therapy
New types of treatment are being tested in clinical trials.
Treatment for childhood carcinoma of unknown primary may cause side effects.
Patients may want to think about taking part in a clinical trial.
Patients can enter clinical trials before, during, or after starting their cancer treatment.
Follow-up tests may be needed.
There are different types of treatment for children with carcinoma of unknown primary.
Some treatments are standard (the currently used treatment), and some are being tested in clinical trials. A treatment clinical trial is a research study meant to help improve current treatments or obtain information on new treatments for patients with cancer. When clinical trials show that a new treatment is better than the standard treatment, the new treatment may become the standard treatment.
Because cancer in children is rare, taking part in a clinical trial should be considered. Some clinical trials are open only to patients who have not started treatment.
Children with carcinoma of unknown primary should have their treatment planned by a team of doctors who are experts in treating childhood cancer.
Treatment will be overseen by a pediatric oncologist, a doctor who specializes in treating children with cancer. The pediatric oncologist works with other pediatric health professionals who are experts in treating children with cancer and who specialize in certain areas of medicine. This may include the following specialists and others:
Chemotherapy is a cancer treatment that uses drugs to stop the growth of cancer cells, either by killing the cells or by stopping them from dividing. When chemotherapy is taken by mouth or injected into a vein or muscle, the drugs enter the bloodstream and can reach cancer cells throughout the body (systemic chemotherapy).
Targeted therapy
Targeted therapy is a treatment that uses drugs or other substances to identify and attack cancer cells.Targeted therapies usually cause less harm to normal cells than chemotherapy and radiation therapy do.
New types of treatment are being tested in clinical trials.
Patients may want to think about taking part in a clinical trial.
For some patients, taking part in a clinical trial may be the best treatment choice. Clinical trials are part of the cancer research process. Clinical trials are done to find out if new cancer treatments are safe and effective or better than the standard treatment.
Many of today’s standard treatments for cancer are based on earlier clinical trials. Patients who take part in a clinical trial may receive the standard treatment or be among the first to receive a new treatment.
Patients who take part in clinical trials also help improve the way cancer will be treated in the future. Even when clinical trials do not lead to effective new treatments, they often answer important questions and help move research forward.
Patients can enter clinical trials before, during, or after starting their cancer treatment.
Some clinical trials only include patients who have not yet received treatment. Other trials test treatments for patients whose cancer has not gotten better. There are also clinical trials that test new ways to stop cancer from recurring (coming back) or reduce the side effects of cancer treatment.
Clinical trials are taking place in many parts of the country. Information about clinical trials supported by NCI can be found on NCI’s clinical trials search webpage. Clinical trials supported by other organizations can be found on the ClinicalTrials.gov website.
Follow-up tests may be needed.
As your child goes through treatment, they will have follow-up tests or check-ups. Some tests that were done to diagnose or stage the cancer may be repeated to see how well the treatment is working. Decisions about whether to continue, change, or stop treatment may be based on the results of these tests.
Some of the tests will continue to be done from time to time after treatment has ended. The results of these tests can show if your child’s condition has changed or if the cancer has recurred (come back).
Treatment of Childhood Carcinoma of Unknown Primary
Sometimes childhood carcinoma of unknown primary can recur (come back) after treatment. If your child is diagnosed with a recurrent carcinoma of unknown primary, your child’s doctor will work with you to plan treatment.
Use our clinical trial search to find NCI-supported cancer clinical trials that are accepting patients. You can search for trials based on the type of cancer, the age of the patient, and where the trials are being done. General information about clinical trials is also available.
To Learn More About Childhood Carcinoma of Unknown Primary
Physician Data Query (PDQ) is the National Cancer Institute’s (NCI’s) comprehensive cancer information database. The PDQ database contains summaries of the latest published information on cancer prevention, detection, genetics, treatment, supportive care, and complementary and alternative medicine. Most summaries come in two versions. The health professional versions have detailed information written in technical language. The patient versions are written in easy-to-understand, nontechnical language. Both versions have cancer information that is accurate and up to date and most versions are also available in Spanish.
PDQ is a service of the NCI. The NCI is part of the National Institutes of Health (NIH). NIH is the federal government’s center of biomedical research. The PDQ summaries are based on an independent review of the medical literature. They are not policy statements of the NCI or the NIH.
Purpose of This Summary
This PDQ cancer information summary has current information about the treatment of childhood carcinoma of unknown primary. It is meant to inform and help patients, families, and caregivers. It does not give formal guidelines or recommendations for making decisions about health care.
Reviewers and Updates
Editorial Boards write the PDQ cancer information summaries and keep them up to date. These Boards are made up of experts in cancer treatment and other specialties related to cancer. The summaries are reviewed regularly and changes are made when there is new information. The date on each summary (“Updated”) is the date of the most recent change.
The information in this patient summary was taken from the health professional version, which is reviewed regularly and updated as needed, by the PDQ Pediatric Treatment Editorial Board.
Clinical Trial Information
A clinical trial is a study to answer a scientific question, such as whether one treatment is better than another. Trials are based on past studies and what has been learned in the laboratory. Each trial answers certain scientific questions in order to find new and better ways to help cancer patients. During treatment clinical trials, information is collected about the effects of a new treatment and how well it works. If a clinical trial shows that a new treatment is better than one currently being used, the new treatment may become “standard.” Patients may want to think about taking part in a clinical trial. Some clinical trials are open only to patients who have not started treatment.
Clinical trials can be found online at NCI’s website. For more information, call the Cancer Information Service (CIS), NCI’s contact center, at 1-800-4-CANCER (1-800-422-6237).
Permission to Use This Summary
PDQ is a registered trademark. The content of PDQ documents can be used freely as text. It cannot be identified as an NCI PDQ cancer information summary unless the whole summary is shown and it is updated regularly. However, a user would be allowed to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks in the following way: [include excerpt from the summary].”
Images in this summary are used with permission of the author(s), artist, and/or publisher for use in the PDQ summaries only. If you want to use an image from a PDQ summary and you are not using the whole summary, you must get permission from the owner. It cannot be given by the National Cancer Institute. Information about using the images in this summary, along with many other images related to cancer can be found in Visuals Online. Visuals Online is a collection of more than 3,000 scientific images.
Disclaimer
The information in these summaries should not be used to make decisions about insurance reimbursement. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s E-mail Us.
Cancers of unknown primary sites present as metastatic cancers for which precise primary tumor sites cannot be determined.[1] As an example, lymph nodes at the base of the skull may enlarge in relationship to a tumor on the face or scalp that is not evident by physical examination or radiographic imaging. Thus, modern imaging techniques may indicate the extent of the disease but not a primary site. Tumors such as adenocarcinomas, melanomas, and embryonal tumors, like rhabdomyosarcomas and neuroblastomas, may present in this way.
Less than 1% of all solid cancers of unknown primary sites occur in children. Because of the age-related incidence of tumor types, embryonal histologies are more common in children.[2]
References
Kuttesch JF, Parham DM, Kaste SC, et al.: Embryonal malignancies of unknown primary origin in children. Cancer 75 (1): 115-21, 1995. [PUBMED Abstract]
Pavlidis N, Pentheroudakis G: Cancer of unknown primary site. Lancet 379 (9824): 1428-35, 2012. [PUBMED Abstract]
Diagnostic Evaluation
For all patients who present with tumors from unknown primary sites, treatment is directed toward the specific histopathology of the tumor and is age-appropriate for the general type of cancer suspected, regardless of the sites involved.[1]
Studies in adults suggest that positron emission tomography (PET) imaging can be helpful in identifying cancers of unknown primary sites, particularly in patients whose tumors arise in the head and neck area.[2] A report in adults using fluorine F 18-fludeoxyglucose PET-computed tomography identified 42.5% of primary tumors in a group of cancers of unknown primary sites.[3]
The use of gene expression profiling and next-generation sequencing can enhance the ability to identify the putative tissue of origin and guide the selection of targeted agents for specific variants.[4–8]
In a study of 200 adult patients with carcinomas of unknown primary sites, 125 had adenocarcinomas and 75 had carcinomas without features of adenocarcinoma. Genomic alterations were found in 96% of the cases. The most common alterations were TP53 (55%), KRAS (20%), CDKN2A (19%), and MYC (12%). Clinically relevant and potentially actionable variants included KRAS (20%), CDKN2A (19%), MCL1 (10%), PTEN (7%), PIK3CA (9%), ERBB2 (8%), RICTOR (6%), BRAF (6%), and NF1 (4%). These findings suggest that genomic profiling can help identify potentially actionable targets, which could benefit patients clinically while reducing the complex, costly workup needed to search for a primary tumor site of origin.[9]
Despite reports of precision medicine studies in pediatric oncology, none of them have described a case of cancer of unknown primary site with a defined or actionable genomic alteration.[10]
References
Kuttesch JF, Parham DM, Kaste SC, et al.: Embryonal malignancies of unknown primary origin in children. Cancer 75 (1): 115-21, 1995. [PUBMED Abstract]
Bohuslavizki KH, Klutmann S, Kröger S, et al.: FDG PET detection of unknown primary tumors. J Nucl Med 41 (5): 816-22, 2000. [PUBMED Abstract]
Han A, Xue J, Hu M, et al.: Clinical value of 18F-FDG PET-CT in detecting primary tumor for patients with carcinoma of unknown primary. Cancer Epidemiol 36 (5): 470-5, 2012. [PUBMED Abstract]
Tothill RW, Li J, Mileshkin L, et al.: Massively-parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. J Pathol 231 (4): 413-23, 2013. [PUBMED Abstract]
Varadhachary GR, Talantov D, Raber MN, et al.: Molecular profiling of carcinoma of unknown primary and correlation with clinical evaluation. J Clin Oncol 26 (27): 4442-8, 2008. [PUBMED Abstract]
Fizazi K, Greco FA, Pavlidis N, et al.: Cancers of unknown primary site: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 26 (Suppl 5): v133-8, 2015. [PUBMED Abstract]
Greco FA, Lennington WJ, Spigel DR, et al.: Poorly differentiated neoplasms of unknown primary site: diagnostic usefulness of a molecular cancer classifier assay. Mol Diagn Ther 19 (2): 91-7, 2015. [PUBMED Abstract]
Gatalica Z, Millis SZ, Vranic S, et al.: Comprehensive tumor profiling identifies numerous biomarkers of drug response in cancers of unknown primary site: analysis of 1806 cases. Oncotarget 5 (23): 12440-7, 2014. [PUBMED Abstract]
Ross JS, Wang K, Gay L, et al.: Comprehensive Genomic Profiling of Carcinoma of Unknown Primary Site: New Routes to Targeted Therapies. JAMA Oncol 1 (1): 40-9, 2015. [PUBMED Abstract]
Mody RJ, Prensner JR, Everett J, et al.: Precision medicine in pediatric oncology: Lessons learned and next steps. Pediatr Blood Cancer 64 (3): , 2017. [PUBMED Abstract]
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has slowly increased since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation to achieve optimal survival and quality of life:
Primary care physicians.
Pediatric surgeons.
Pathologists.
Pediatric radiation oncologists.
Pediatric medical oncologists and hematologists.
Ophthalmologists.
Rehabilitation specialists.
Pediatric oncology nurses.
Social workers.
Child-life professionals.
Psychologists.
Nutritionists.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children and adolescents with cancer.[2] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Other types of clinical trials test novel therapies when there is no standard therapy for a cancer diagnosis. Most of the progress in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[3–5] Childhood and adolescent cancer survivors require close monitoring because side effects of cancer therapy may persist or develop months or years after treatment. For information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
Childhood cancer is a rare disease, with about 15,000 cases diagnosed annually in the United States in individuals younger than 20 years.[6] The U.S. Rare Diseases Act of 2002 defines a rare disease as one that affects populations smaller than 200,000 people in the United States. Therefore, all pediatric cancers are considered rare.
The designation of a rare tumor is not uniform among pediatric and adult groups. In adults, rare cancers are defined as those with an annual incidence of fewer than six cases per 100,000 people. They account for up to 24% of all cancers diagnosed in the European Union and about 20% of all cancers diagnosed in the United States.[7,8] In children and adolescents, the designation of a rare tumor is not uniform among international groups, as follows:
A consensus effort between the European Union Joint Action on Rare Cancers and the European Cooperative Study Group for Rare Pediatric Cancers estimated that 11% of all cancers in patients younger than 20 years could be categorized as very rare. This consensus group defined very rare cancers as those with annual incidences of fewer than two cases per 1 million people. However, three additional histologies (thyroid carcinoma, melanoma, and testicular cancer) with incidences of more than two cases per 1 million people were also included in the very rare group due to a lack of knowledge and expertise in the management of these tumors.[9]
The Children’s Oncology Group defines rare pediatric cancers as those listed in the International Classification of Childhood Cancer subgroup XI, which includes thyroid cancers, melanomas and nonmelanoma skin cancers, and multiple types of carcinomas (e.g., adrenocortical carcinomas, nasopharyngeal carcinomas, and most adult-type carcinomas such as breast cancers and colorectal cancers).[10] These diagnoses account for about 5% of the cancers diagnosed in children aged 0 to 14 years and about 27% of the cancers diagnosed in adolescents aged 15 to 19 years.[4]
Most cancers in subgroup XI are either melanomas or thyroid cancers, with other cancer types accounting for only 2% of the cancers diagnosed in children aged 0 to 14 years and 9.3% of the cancers diagnosed in adolescents aged 15 to 19 years.
These rare cancers are extremely challenging to study because of the relatively few patients with any individual diagnosis, the predominance of rare cancers in the adolescent population, and the small number of clinical trials for adolescents with rare cancers.
Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010. [PUBMED Abstract]
American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed February 25, 2025.
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.
Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr. [PUBMED Abstract]
Gatta G, Capocaccia R, Botta L, et al.: Burden and centralised treatment in Europe of rare tumours: results of RARECAREnet-a population-based study. Lancet Oncol 18 (8): 1022-1039, 2017. [PUBMED Abstract]
DeSantis CE, Kramer JL, Jemal A: The burden of rare cancers in the United States. CA Cancer J Clin 67 (4): 261-272, 2017. [PUBMED Abstract]
Ferrari A, Brecht IB, Gatta G, et al.: Defining and listing very rare cancers of paediatric age: consensus of the Joint Action on Rare Cancers in cooperation with the European Cooperative Study Group for Pediatric Rare Tumors. Eur J Cancer 110: 120-126, 2019. [PUBMED Abstract]
Pappo AS, Krailo M, Chen Z, et al.: Infrequent tumor initiative of the Children’s Oncology Group: initial lessons learned and their impact on future plans. J Clin Oncol 28 (33): 5011-6, 2010. [PUBMED Abstract]
Treatment of Childhood Cancer of Unknown Primary
Chemotherapy, targeted therapy, and radiation therapy may be used to treat childhood cancers of unknown primary sites. The appropriate and relevant treatments, according to the general category of carcinoma or sarcoma (depending on the histological findings, symptoms, and extent of tumor), are initiated as early as possible.[1]
Morris GJ, Greco FA, Hainsworth JD, et al.: Cancer of unknown primary site. Semin Oncol 37 (2): 71-9, 2010. [PUBMED Abstract]
Treatment Options Under Clinical Evaluation for Childhood Cancer of Unknown Primary
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Latest Updates to This Summary (08/13/2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
This summary was comprehensively reviewed.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of pediatric cancer of unknown primary. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Cancer of Unknown Primary (CUP) Treatment are:
Denise Adams, MD (Children’s Hospital Boston)
Karen J. Marcus, MD, FACR (Dana-Farber of Boston Children’s Cancer Center and Blood Disorders Harvard Medical School)
William H. Meyer, MD
Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children’s Healthcare of Atlanta – Egleston Campus)
Alberto S. Pappo, MD (St. Jude Children’s Research Hospital)
Arthur Kim Ritchey, MD (Children’s Hospital of Pittsburgh of UPMC)
Carlos Rodriguez-Galindo, MD (St. Jude Children’s Research Hospital)
Stephen J. Shochat, MD (St. Jude Children’s Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Cancer of Unknown Primary (CUP) Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/unknown-primary/hp/child-unknown-primary-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 31909936]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
The only widely proposed screening procedure for skin cancer is visual examination of the skin, including both self-examination by the patient and clinical examination by the health care provider. Mobile phone applications that evaluate skin lesions to detect skin cancer and malignant melanoma have been launched.[1] However, the use of such applications to assess skin cancer has been problematic because of the lack of evidence of their diagnostic accuracy and because they have not been studied in large-scale screening programs.[2–4] The use of convolutional neural networks to classify images of melanoma and skin cancer is a growing area of research.[5–7]
Benefits
There is insufficient evidence that population screening for skin cancer reduces skin cancer mortality. The evidence is inadequate to determine whether visual examination of the skin in asymptomatic individuals leads to a reduction in mortality from melanomatous skin cancer. Further, in asymptomatic populations, the effect of visual skin examination on mortality from nonmelanomatous skin cancers is unknown.
Magnitude of Effect: Unknown.
Study Design: Direct evidence limited to a single ecological study.
Internal Validity: Poor.
Consistency: Not applicable.
External Validity: Poor.
Harms
Based on fair—though unquantified—evidence, visual examination of the skin in asymptomatic individuals may lead to adverse consequences. These consequences include complications of diagnostic or treatment interventions (such as poor cosmetic or functional outcomes) and the psychological effects of being labeled with a potentially fatal disease. Other harmful consequences are overdiagnosis, leading to the detection of biologically benign disease that would otherwise go undetected, and possible misdiagnosis of a benign lesion as malignant.
Magnitude of Effect: Unknown.
Study Design: Case series, ecological studies.
Internal Validity: Fair.
Consistency: Fair.
External Validity: Fair.
References
Buechi R, Faes L, Bachmann LM, et al.: Evidence assessing the diagnostic performance of medical smartphone apps: a systematic review and exploratory meta-analysis. BMJ Open 7 (12): e018280, 2017. [PUBMED Abstract]
Kassianos AP, Emery JD, Murchie P, et al.: Smartphone applications for melanoma detection by community, patient and generalist clinician users: a review. Br J Dermatol 172 (6): 1507-1518, 2015. [PUBMED Abstract]
Wolf JA, Moreau JF, Akilov O, et al.: Diagnostic inaccuracy of smartphone applications for melanoma detection. JAMA Dermatol 149 (4): 422-6, 2013. [PUBMED Abstract]
Udrea A, Mitra GD, Costea D, et al.: Accuracy of a smartphone application for triage of skin lesions based on machine learning algorithms. J Eur Acad Dermatol Venereol 34 (3): 648-655, 2020. [PUBMED Abstract]
Hekler A, Utikal JS, Enk AH, et al.: Superior skin cancer classification by the combination of human and artificial intelligence. Eur J Cancer 120: 114-121, 2019. [PUBMED Abstract]
Esteva A, Kuprel B, Novoa RA, et al.: Dermatologist-level classification of skin cancer with deep neural networks. Nature 542 (7639): 115-118, 2017. [PUBMED Abstract]
Phillips M, Marsden H, Jaffe W, et al.: Assessment of Accuracy of an Artificial Intelligence Algorithm to Detect Melanoma in Images of Skin Lesions. JAMA Netw Open 2 (10): e1913436, 2019. [PUBMED Abstract]
Incidence and Mortality of Skin Cancer
There are two main types of skin cancer:
Keratinocyte carcinoma.
Basal cell carcinoma (BCC).
Squamous cell carcinoma (SCC).
Melanoma.
BCC and SCC are the most common forms of skin cancer but have substantially better prognoses than the less common, generally more aggressive melanoma.
Keratinocyte carcinoma is the most commonly occurring cancer in the United States. Its incidence appears to be increasing in some [1] but not all [2] areas of the United States. Overall U.S. incidence rates have likely been increasing for a number of years.[3,4] At least some of this increase may be attributable to increased skin cancer awareness and resultant increasing investigation and biopsy of skin lesions. A precise estimate of the total number and incidence rate of keratinocyte carcinoma is not possible because reporting to cancer registries is not required. However, it was estimated that in 2012, 5.4 million cases of keratinocyte carcinoma were diagnosed among 3.3 million people in the United States.[5] That number exceeds all other cases of cancer estimated by the American Cancer Society for 2025, which is about 2 million.[5]
Melanoma is reportable in U.S. cancer registries, so there are more reliable estimates of incidence than for keratinocyte carcinoma. In 2025, it is estimated that 104,960 individuals in the United States will be diagnosed with invasive melanoma and 107,240 will be diagnosed with melanoma in situ. Approximately 8,430 individuals will die of melanoma in 2025. Since the early 2000s, melanoma incidence rates among individuals younger than 50 years have stabilized in women but declined by about 1% per year in men. However, among individuals aged 50 years and older in recent years, the incidence rates appeared to have stabilized in men but increased by about 3% per year in women.[5] From 2013 to 2022, melanoma mortality rates declined by about 4% per year and 3% per year in men and women, respectively.[5]
A study of skin biopsy rates in relation to melanoma incidence rates obtained from the Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute indicated that much of the observed increase in incidence between 1986 and 2001 was confined to local disease and was most likely caused by overdiagnosis as a result of increased skin biopsy rates during this period.[6] A second study that used SEER data between 2002 and 2009 reported similar findings.[7]
The incidence of melanoma also increased in children and adolescents until 2001. However, between 2001 and 2022, there was a 4.3% reduction in the yearly incidence rate of melanoma among children and adolescents in the National Childhood Cancer Registry databases.[8] During that time, the average annual incidence in this group was exceptionally low (4.5 per 1 million), which may have resulted in spurious trends.[8] Nevertheless, similar trends have been seen in Sweden.[9] In the U.S. study of pediatric melanoma, nearly one-half of the patients had local disease (22% of patients had in situ disease, and 25% of patients had superficial spreading), and nearly one-half of the patients had disease with a thickness of less than one millimeter. Given that mortality from pediatric melanoma had been fairly stable during those years,[10] it is likely that the increase in incidence could be explained, at least in part, by overdiagnosis.
References
Athas WF, Hunt WC, Key CR: Changes in nonmelanoma skin cancer incidence between 1977-1978 and 1998-1999 in Northcentral New Mexico. Cancer Epidemiol Biomarkers Prev 12 (10): 1105-8, 2003. [PUBMED Abstract]
Harris RB, Griffith K, Moon TE: Trends in the incidence of nonmelanoma skin cancers in southeastern Arizona, 1985-1996. J Am Acad Dermatol 45 (4): 528-36, 2001. [PUBMED Abstract]
Rogers HW, Weinstock MA, Harris AR, et al.: Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol 146 (3): 283-7, 2010. [PUBMED Abstract]
Leiter U, Eigentler T, Garbe C: Epidemiology of skin cancer. Adv Exp Med Biol 810: 120-40, 2014. [PUBMED Abstract]
American Cancer Society: Cancer Facts and Figures 2025. American Cancer Society, 2025. Available online. Last accessed January 16, 2025.
Welch HG, Woloshin S, Schwartz LM: Skin biopsy rates and incidence of melanoma: population based ecological study. BMJ 331 (7515): 481, 2005. [PUBMED Abstract]
Weinstock MA, Lott JP, Wang Q, et al.: Skin biopsy utilization and melanoma incidence among Medicare beneficiaries. Br J Dermatol 176 (4): 949-954, 2017. [PUBMED Abstract]
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
Austin MT, Xing Y, Hayes-Jordan AA, et al.: Melanoma incidence rises for children and adolescents: an epidemiologic review of pediatric melanoma in the United States. J Pediatr Surg 48 (11): 2207-13, 2013. [PUBMED Abstract]
Lewis KG: Trends in pediatric melanoma mortality in the United States, 1968 through 2004. Dermatol Surg 34 (2): 152-9, 2008. [PUBMED Abstract]
Risk Factors for Skin Cancer
Epidemiological evidence suggests that exposure to UV radiation and the sensitivity of an individual’s skin to UV radiation are risk factors for skin cancer, although the type of exposure (high-intensity and short-duration vs. chronic exposure) and pattern of exposure (continuous vs. intermittent) may differ among the three main types of skin cancer.[1–3] In addition, genetic predisposition and the immune system may play roles in the pathogenesis of skin cancers.[4] Organ-transplant recipients receiving immunosuppressive drugs are at elevated risk of skin cancers, particularly squamous cell carcinoma (SCC). Arsenic exposure also increases the risk of cutaneous SCC.[5,6]
The incidence of melanoma rises rapidly in White individuals after age 20 years. Fair-skinned individuals exposed to the sun are at higher risk. Individuals with certain types of pigmented lesions (dysplastic or atypical nevi), with several large nondysplastic nevi, many small nevi, or moderate freckling have a twofold to threefold increased risk of developing melanoma.[7] Individuals with familial dysplastic nevus syndrome or with several dysplastic or atypical nevi are at high (>fivefold) risk of developing melanoma.[4,7]
It is important to note that, for the general population, most melanomas may not arise from preexisting nevi. A meta-analysis of studies published between 1948 and 2016 found that the prevalence of nevus-associated melanomas was only 29%, compared with 71% for the prevalence of de novo melanomas.[8]
References
Koh HK: Cutaneous melanoma. N Engl J Med 325 (3): 171-82, 1991. [PUBMED Abstract]
Preston DS, Stern RS: Nonmelanoma cancers of the skin. N Engl J Med 327 (23): 1649-62, 1992. [PUBMED Abstract]
English DR, Armstrong BK, Kricker A, et al.: Case-control study of sun exposure and squamous cell carcinoma of the skin. Int J Cancer 77 (3): 347-53, 1998. [PUBMED Abstract]
Hawkes JE, Truong A, Meyer LJ: Genetic predisposition to melanoma. Semin Oncol 43 (5): 591-597, 2016. [PUBMED Abstract]
Thomas VD, Aasi SZ, Wilson LD, et al.: Cancer of the skin. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds.: Cancer: Principles and Practice of Oncology. Vols. 1 & 2. 8th ed. Lippincott Williams & Wilkins, 2008, pp 1863-87.
Le Mire L, Hollowood K, Gray D, et al.: Melanomas in renal transplant recipients. Br J Dermatol 154 (3): 472-7, 2006. [PUBMED Abstract]
Gandini S, Sera F, Cattaruzza MS, et al.: Meta-analysis of risk factors for cutaneous melanoma: I. Common and atypical naevi. Eur J Cancer 41 (1): 28-44, 2005. [PUBMED Abstract]
Pampena R, Kyrgidis A, Lallas A, et al.: A meta-analysis of nevus-associated melanoma: Prevalence and practical implications. J Am Acad Dermatol 77 (5): 938-945.e4, 2017. [PUBMED Abstract]
Accuracy of Making a Clinical Diagnosis of Melanoma
Observer variability among physicians has been noted in the evaluation of skin lesions and subsequent biopsy specimens. A systematic review of 32 studies that compared the accuracy of dermatologists and primary care physicians in making a clinical diagnosis of melanoma concluded that there was no statistically significant difference in accuracy. However, the results were inconclusive, owing to small sample sizes and study design weaknesses.[1] Subsequent studies have noted a higher accuracy for dermatologists in the diagnosis of melanocytic lesions,[2,3] yet there is a shortage of dermatologists to meet the demands of population-level screening.
A study of 187 pathologists who practiced in the United States found that cases of moderately dysplastic nevi to early-stage invasive melanoma had less than 50% agreement with a reference diagnosis defined by consensus of experienced pathologists.[4] At a U.S. population level, it is estimated that 82.8% (95% confidence interval, 81.0%–84.5%) of melanocytic skin biopsy diagnoses would be verified if they were reviewed by a consensus reference panel of experienced pathologists.[4] In addition, differentiating between benign and malignant melanocytic tumors during histological examinations of biopsy specimens has been shown to be inconsistent, even in the hands of experienced dermatopathologists.[5,6] This variability in the diagnosis of melanocytic lesions undermines the results of studies that examine screening effectiveness and also may undermine the effectiveness of any screening intervention. Furthermore, this finding suggests that requesting a second opinion regarding the pathology of biopsy specimens may be important.[5–7] A standardized approach to pathologists’ classifying of the interpretations of melanocytic skin lesions may also reduce confusion and improve communication between clinicians.[4,6,8,9]
References
Chen SC, Bravata DM, Weil E, et al.: A comparison of dermatologists’ and primary care physicians’ accuracy in diagnosing melanoma: a systematic review. Arch Dermatol 137 (12): 1627-34, 2001. [PUBMED Abstract]
Chen SC, Pennie ML, Kolm P, et al.: Diagnosing and managing cutaneous pigmented lesions: primary care physicians versus dermatologists. J Gen Intern Med 21 (7): 678-82, 2006. [PUBMED Abstract]
Corbo MD, Wismer J: Agreement between dermatologists and primary care practitioners in the diagnosis of malignant melanoma: review of the literature. J Cutan Med Surg 16 (5): 306-10, 2012 Sep-Oct. [PUBMED Abstract]
Elmore JG, Barnhill RL, Elder DE, et al.: Pathologists’ diagnosis of invasive melanoma and melanocytic proliferations: observer accuracy and reproducibility study. BMJ 357: j2813, 2017. [PUBMED Abstract]
Farmer ER, Gonin R, Hanna MP: Discordance in the histopathologic diagnosis of melanoma and melanocytic nevi between expert pathologists. Hum Pathol 27 (6): 528-31, 1996. [PUBMED Abstract]
Lott JP, Elmore JG, Zhao GA, et al.: Evaluation of the Melanocytic Pathology Assessment Tool and Hierarchy for Diagnosis (MPATH-Dx) classification scheme for diagnosis of cutaneous melanocytic neoplasms: Results from the International Melanoma Pathology Study Group. J Am Acad Dermatol 75 (2): 356-63, 2016. [PUBMED Abstract]
Piepkorn MW, Longton GM, Reisch LM, et al.: Assessment of Second-Opinion Strategies for Diagnoses of Cutaneous Melanocytic Lesions. JAMA Netw Open 2 (10): e1912597, 2019. [PUBMED Abstract]
Piepkorn MW, Barnhill RL, Elder DE, et al.: The MPATH-Dx reporting schema for melanocytic proliferations and melanoma. J Am Acad Dermatol 70 (1): 131-41, 2014. [PUBMED Abstract]
Radick AC, Reisch LM, Shucard HL, et al.: Terminology for melanocytic skin lesions and the MPATH-Dx classification schema: A survey of dermatopathologists. J Cutan Pathol 48 (6): 733-738, 2021. [PUBMED Abstract]
Evidence of Benefit Associated With Screening
More than 90% of melanomas that arise in the skin can be recognized with the naked eye. Very often there is a prolonged horizontal growth phase, during which the tumor expands centrifugally beneath the epidermis but does not invade the underlying dermis. This horizontal growth phase may provide lead time for early detection. Melanoma is more easily cured if treated before the onset of the vertical growth phase with its metastatic potential.[1]
The probability of tumor recurrence within 10 years after curative resection is less than 10% with tumors less than 1.4 mm in thickness. For patients with tumors less than 0.76 mm in thickness, the likelihood of recurrence is less than 1% in 10 years.[2]
A systematic review of skin cancer screening examined evidence available through mid-2005. The review concluded that direct evidence of improved health outcomes associated with skin cancer screening is lacking.[3] An updated review published in 2016 found limited evidence that skin cancer screening reduces melanoma mortality.[4,5]
No randomized trials evaluating the efficacy of skin cancer screening on mortality have been completed. A population-based trial (using cluster randomization) to determine the effect of skin cancer screening on melanoma mortality was initiated in Queensland, Australia, but lost its funding after the initial pilot phase, and no health outcomes were ever reported.[6]
Two ecological studies have been conducted using data from Germany. The first study was a pilot project conducted in 2003 and 2004, in which a skin cancer screening program was implemented in one federal state. Suggestion of a reduction in melanoma mortality with screening led to the establishment of countywide skin cancer screening programs in 2008.[7,8] The programs offered a whole-body skin examination once every 2 years for individuals older than 35 years. The second ecological study compared the melanoma mortality experience in Germany with the melanoma mortality experience of subregions of 22 European countries—none of which had organized screening programs—for the years 2000 to 2013. After adjustment for potential confounders, Germany and the 22 European regions had similar malignant mortality rates, suggesting no benefit of screening.[9]
References
Friedman RJ, Rigel DS, Kopf AW: Early detection of malignant melanoma: the role of physician examination and self-examination of the skin. CA Cancer J Clin 35 (3): 130-51, 1985 May-Jun. [PUBMED Abstract]
Blois MS, Sagebiel RW, Abarbanel RM, et al.: Malignant melanoma of the skin. I. The association of tumor depth and type, and patient sex, age, and site with survival. Cancer 52 (7): 1330-41, 1983. [PUBMED Abstract]
Wolff T, Tai E, Miller T: Screening for skin cancer: an update of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 150 (3): 194-8, 2009. [PUBMED Abstract]
Wernli KJ, Henrikson NB, Morrison CC, et al.: Screening for Skin Cancer in Adults: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 316 (4): 436-47, 2016. [PUBMED Abstract]
Bibbins-Domingo K, Grossman DC, Curry SJ, et al.: Screening for Skin Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 316 (4): 429-35, 2016. [PUBMED Abstract]
Aitken JF, Elwood JM, Lowe JB, et al.: A randomised trial of population screening for melanoma. J Med Screen 9 (1): 33-7, 2002. [PUBMED Abstract]
Katalinic A, Waldmann A, Weinstock MA, et al.: Does skin cancer screening save lives? An observational study comparing trends in melanoma mortality in regions with and without screening. Cancer 118 (21): 5395-402, 2012. [PUBMED Abstract]
Eisemann N, Waldmann A, Holleczek B, et al.: Observed and expected mortality in the German skin cancer screening pilot project SCREEN. J Med Screen 25 (3): 166-168, 2018. [PUBMED Abstract]
Kaiser M, Schiller J, Schreckenberger C: The effectiveness of a population-based skin cancer screening program: evidence from Germany. Eur J Health Econ 19 (3): 355-367, 2018. [PUBMED Abstract]
Evidence of Harms Associated With Screening
Harms have not been well studied or reported in quantitative terms, but the potential for adverse consequences from skin cancer screening exists. In the SCREEN pilot project in Germany, 4.4% of all screened participants underwent a skin excision for a suspicious lesion, but most biopsies did not result in a cancer diagnosis. The detection rate was especially affected by age. One case of melanoma was detected per 28 excisions overall (for both men and women), while 52 skin excisions were required to detect one melanoma in men aged 20 to 34 years.[1]
Visual examination of the skin in asymptomatic individuals may lead to cosmetic or functional complications of diagnostic or treatment interventions and psychological effects of being labeled with a potentially fatal disease. However, robust data on the frequency of such events are lacking. Other harmful consequences are overdiagnosis, leading to the detection of biologically benign disease that would otherwise go undetected,[2–4] and possible misdiagnosis of a benign lesion as malignant. For more information, see the Accuracy of Making a Clinical Diagnosis of Melanoma section.
References
Waldmann A, Nolte S, Geller AC, et al.: Frequency of excisions and yields of malignant skin tumors in a population-based screening intervention of 360,288 whole-body examinations. Arch Dermatol 148 (8): 903-10, 2012. [PUBMED Abstract]
Welch HG, Woloshin S, Schwartz LM: Skin biopsy rates and incidence of melanoma: population based ecological study. BMJ 331 (7515): 481, 2005. [PUBMED Abstract]
Weinstock MA, Lott JP, Wang Q, et al.: Skin biopsy utilization and melanoma incidence among Medicare beneficiaries. Br J Dermatol 176 (4): 949-954, 2017. [PUBMED Abstract]
Schoffer O, Schülein S, Arand G, et al.: Tumour stage distribution and survival of malignant melanoma in Germany 2002-2011. BMC Cancer 16 (1): 936, 2016. [PUBMED Abstract]
Latest Updates to This Summary (05/09/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Updated statistics with estimated new cases of melanoma and melanoma in situ and deaths due to melanoma for 2025. Also revised text to state that from 2013 to 2022, melanoma mortality rates declined by about 4% per year and 3% per year in men and women, respectively.
This summary is written and maintained by the PDQ Screening and Prevention Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about skin cancer screening. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Screening and Prevention Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Screening and Prevention Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Screening and Prevention Editorial Board. PDQ Skin Cancer Screening. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/skin/hp/skin-screening-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389300]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
The information in these summaries should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Melanoma of the uveal tract (iris, ciliary body, and choroid) is rare, but it is the most common primary intraocular malignancy in adults. The mean age-adjusted incidence of uveal melanoma in the United States is approximately 4.3 new cases per million people, with no clear variation by latitude. The incidence is higher in men (4.9 cases per million) than in women (3.7 cases per million).[1] The age-adjusted incidence of this cancer has remained stable since at least the early 1970s.[1,2] U.S. incidence rates are lower than the rates of other reporting countries, which vary from about 5.3 to 10.9 cases per million. Some of the variation may be the result of differences in inclusion criteria and methods of calculation.[1]
Uveal melanoma is most often diagnosed in older individuals, with a progressively rising, age-specific incidence rate that peaks near age 70 years.[3]
Host susceptibility factors associated with the development of this cancer include:[2–4]
White race and ethnicity.
Light eye color.
Fair skin.
The ability to tan.
In view of these susceptibility factors, numerous observational studies have explored the relationship between sunlight exposure and risk of uveal melanoma. These studies have found only weak associations or yielded contradictory results.[3] Similarly, there is no consistent evidence that occupational exposure to UV light or other agents is a risk factor for uveal melanoma.[3,5]
Anatomy
Uveal melanomas can arise in the anterior (iris) or the posterior (ciliary body or choroid) uveal tract.[6] Most uveal tract melanomas originate in the choroid. The ciliary body is a less common site of origin, and the iris is the least common. The comparatively low incidence of iris melanomas has been attributed to the characteristic features of these tumors; they tend to be smaller, slower growing, and relatively dormant compared with their posterior counterparts. Iris melanomas rarely metastasize.[7] Melanomas of the posterior uveal tract generally have a more malignant histological appearance; are detected later; and metastasize more frequently than iris melanomas. The typical choroidal melanoma is a brown, elevated, dome-shaped subretinal mass. The degree of pigmentation ranges from dark brown to totally amelanotic.
Most uveal melanomas are initially completely asymptomatic. As the tumor enlarges, it may cause distortion of the pupil (iris melanoma), blurred vision (ciliary body melanoma), or markedly decreased visual acuity caused by secondary retinal detachment (choroidal melanoma). Serous detachment of the retina may occur. If extensive detachment occurs, secondary angle-closure glaucoma occasionally develops. Clinically, several lesions simulate uveal melanoma, including metastatic carcinoma, posterior scleritis, and benign tumors, such as nevi and hemangiomas.[8]
Careful examination by an experienced clinician remains the most important test to diagnose intraocular melanoma. A small uveal melanoma cannot be distinguished from a nevus. Small uveal lesions are observed for growth before making a diagnosis of melanoma. Clinical findings that may help to identify melanoma include:[6]
Orange pigment on the tumor surface.
Subretinal fluid.
Tumor thickness of more than 2 mm.
Low internal reflectivity on ultrasound examination.
Ancillary diagnostic testing, including fluorescein angiography and ultrasonography, can be extremely valuable in establishing and confirming the diagnosis.[9] In a large, retrospective, single-center series of 2,514 consecutive patients with choroidal nevi, the progression rate to melanoma was 8.6% at 5 years, 12.8% at 10 years, and 17.3% at 15 years.[10]
Prognostic Factors
Several factors influence prognosis. The most important factors include:
Several additional microscopic features can affect the prognosis of intraocular melanoma, including:
Mitotic activity.
Lymphocytic infiltration.
Fibrovascular loops (possibly).
Cell type is the most commonly used predictor of outcome following enucleation. Patients with spindle-A cell melanomas have the best prognosis and patients with epithelioid cell melanomas have the least favorable prognosis.[1,4,9] Nevertheless, most tumors have an admixture of cell types, and there is no clear consensus regarding the proportion of epithelioid cells that constitutes designation of a tumor as mixed or epithelioid.[6]
Extraocular extension, recurrence, and metastasis are associated with an extremely poor prognosis, and long-term survival cannot be expected for patients with these features.[11] The 5-year mortality rate for patients with metastasis from ciliary body or choroidal melanoma is approximately 30%, compared with a rate of 2% to 3% for patients with iris melanomas.[12]
References
Singh AD, Topham A: Incidence of uveal melanoma in the United States: 1973-1997. Ophthalmology 110 (5): 956-61, 2003. [PUBMED Abstract]
Inskip PD, Devesa SS, Fraumeni JF: Trends in the incidence of ocular melanoma in the United States, 1974-1998. Cancer Causes Control 14 (3): 251-7, 2003. [PUBMED Abstract]
Weis E, Shah CP, Lajous M, et al.: The association between host susceptibility factors and uveal melanoma: a meta-analysis. Arch Ophthalmol 124 (1): 54-60, 2006. [PUBMED Abstract]
Harris RB, Griffith K, Moon TE: Trends in the incidence of nonmelanoma skin cancers in southeastern Arizona, 1985-1996. J Am Acad Dermatol 45 (4): 528-36, 2001. [PUBMED Abstract]
Albert DM, Kulkarni AD: Intraocular melanoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 2090-8.
Shields CL, Furuta M, Berman EL, et al.: Choroidal nevus transformation into melanoma: analysis of 2514 consecutive cases. Arch Ophthalmol 127 (8): 981-7, 2009. [PUBMED Abstract]
Gragoudas ES, Egan KM, Seddon JM, et al.: Survival of patients with metastases from uveal melanoma. Ophthalmology 98 (3): 383-9; discussion 390, 1991. [PUBMED Abstract]
Introduction to melanocytic tumors of the uvea. In: Shields JA, Shields CL: Intraocular Tumors: A Text and Atlas. Saunders, 1992, pp 45-59.
Cellular Classification of Intraocular (Uveal) Melanoma
Primary intraocular melanomas originate from melanocytes in the uveal tract.[1] The following four distinct cellular types are recognized in intraocular melanoma (revised Callender classification):[2]
Spindle-A cells (spindle-shaped cells with slender nuclei and lacking visible nucleoli).
Spindle-B cells (spindle-shaped cells with larger nuclei and distinct nucleoli).
Epithelioid cells (larger polygonal cells with one or more prominent nucleoli).
Intermediate cells (similar to but smaller than epithelioid cells).
Most primary intraocular melanomas contain variable proportions of epithelioid, spindle-A, and spindle-B cells (mixed-cell melanomas). Pure epithelioid-cell primary melanomas are infrequent (approximately 3% of cases).[1] In the Collaborative Ocular Melanoma Study, mixed-cell melanomas predominated (86% of cases).[3]
References
Klintworth GK, Scroggs MW: The eye and ocular adnexa. In: Sternberg SS, ed.: Diagnostic Surgical Pathology. Lippincott Williams & Wilkins, 1999, pp 994-6.
Grossniklaus HE, Green WR: Uveal tumors. In: Garner A, Klintworth GK, eds.: Pathobiology of Ocular Disease: A Dynamic Approach. 2nd ed. M. Dekker, 1994, pp 1423-77.
Histopathologic characteristics of uveal melanomas in eyes enucleated from the Collaborative Ocular Melanoma Study. COMS report no. 6. Am J Ophthalmol 125 (6): 745-66, 1998. [PUBMED Abstract]
Classification and Stage Information for Intraocular (Uveal) Melanoma
Tumor Size
Uveal melanoma most often assumes a nodular or dome-shaped configuration. Occasionally, tumors are flat or diffuse and involve extensive areas of the uvea with little elevation.
Tumor size classifications according to boundary lines used in a Collaborative Ocular Melanoma Study (COMS) are as follows:[1]
Small: Range from 1.0 to 3.0 mm in apical height and largest basal diameter of 5.0 to 16.0 mm.[1]
Medium: Range from 3.1 to 8.0 mm in apical height and a basal diameter of not more than 16.0 mm.[2]
Large: Greater than 8.0 mm in apical height or a basal diameter more than 16.0 mm, when the apical height is at least 2.0 mm.
Although most ocular melanomas have a raised configuration, about 5% grow in a diffuse pattern that also may have prognostic significance. The tumors have a horizontal, flat-growth pattern, with the thickness measuring approximately 20% or less than the greatest basal dimension. This uncommon variant of uveal melanoma seems to be associated with a poorer prognosis, particularly when the diameter is large and the margins are poorly defined.[3]
In clinical practice, the tumor base may be estimated in average optic disc diameters (1 dd = 1.5 mm). The average elevation may be estimated in diopters (3 diopters = 1 mm). Other techniques, such as ultrasonography, are used to provide more accurate measurements.
An important function of ophthalmic ultrasonography is the detection of extrascleral extension.[4,5] Extrascleral extension measuring 2 mm or more in thickness can be demonstrated, provided it is located behind the equator where the intraocular tumor, sclera, and adjacent orbital fat are readily imaged.[6] Orbital extraocular extension of choroidal melanoma may be found in eyes with medium and large tumors, but it is very rare in eyes with small melanomas.
Metastatic Disease
Systemic metastases are evident in only 2% to 3% of patients at the time of diagnosis of the primary ocular melanoma.[7] Because the uveal tract is a vascular structure without lymphatic channels, tumor spread occurs principally by local extension and by dissemination through the bloodstream.[7] Lymphatic spread is rare but may occur after local extension into the conjunctiva and its lymphatics.[8] Given the rarity of nodal metastases, sentinel node biopsies of nonclinically involved nodes are not done as part of the staging procedure.[7]
Systemic metastases are generally hematogenous in origin, and the first site identified is usually the liver.[9] Lung, bone, and subcutaneous sites are also common.[9] In the COMS trials, the liver was the only site of detectable metastasis in 46% of patients with metastases reported during follow-up or at the time of death; 43% had metastases diagnosed in the liver and other sites.[9] In patients with a history of ocular melanoma who present with hepatic metastases of unknown origin, metastatic melanoma is considered in the differential diagnosis.
It is particularly unusual for choroidal melanomas of any size to invade the optic nerve or its meninges.[10] Metastasis of choroidal melanoma to the contralateral choroid is also rare.[9,11]
Staging
American Joint Committee on Cancer (AJCC) stage groupings and definitions of TNM
The AJCC has designated staging by TNM (tumor, node, metastasis) classification to define melanoma of the uveal tract.[7]
As in the seventh edition of the AJCC Cancer Staging Manual, there is no staging system for iris melanomas in the eighth edition. However, TNM should still be recorded for this site and histology combination.
Table 1. Definition of Primary Tumor (T) for Iris Melanomasa,b
T Category
T Criteria
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
bIris melanomas originate from, and are predominantly located in, this region of the uvea. If less than half the tumor volume is located within the iris, the tumor may have originated in the ciliary body, and consideration should be given to classifying it accordingly.
TX
Primary tumor cannot be assessed.
T0
No evidence of primary tumor.
T1
Tumor limited to the iris.
–T1a
Tumor limited to the iris, not more than 3 clock hours in size.
–T1b
Tumor limited to the iris, more than 3 clock hours in size.
–T1c
Tumor limited to the iris with secondary glaucoma.
T2
Tumor confluent with or extending into the ciliary body, choroid, or both.
–T2a
Tumor confluent with or extending into the ciliary body, without secondary glaucoma.
–T2b
Tumor confluent with or extending into the ciliary body and choroid, without secondary glaucoma.
–T2c
Tumor confluent with or extending into the ciliary body, choroid, or both, with secondary glaucoma.
T3
Tumor confluent with or extending into the ciliary body, choroid, or both, with scleral extension.
T4
Tumor with extrascleral extension.
–T4a
Tumor with extrascleral extension ≤5 mm in largest diameter.
–T4b
Tumor with extrascleral extension >5 mm in largest diameter.
Table 2. Definition of Regional Lymph Node (N)a
N Category
N Criteria
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
NX
Regional lymph nodes cannot be assessed.
N0
No regional lymph node involvement.
N1
Regional lymph node metastases or discrete tumor deposits in the orbit.
–N1a
Metastasis in one or more regional lymph node(s).
–N1b
No regional lymph nodes are positive, but there are discrete tumor deposits in the orbit that are not contiguous to the eye (choroidal and ciliary body).
Table 3. Definition of Distant Metastasis (M)a
M Category
M Criteria
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
M0
No distant metastasis by clinical classification.
M1
Distant metastasis.
–M1a
Largest diameter of the largest metastasis ≤3.0 cm.
–M1b
Largest diameter of the largest metastasis 3.1–8.0 cm.
–M1c
Largest diameter of the largest metastasis ≥8.1 cm.
Table 4. Classification of Ciliary Body and Choroid Uveal Melanoma Based on Thickness and Diametera
Category
Tumor Size
aAdapted from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
1
Tumor is ≤12 mm in diameter and ≤3 mm in thickness; or
Tumor is ≤9 mm in diameter and 3.1–6 mm in thickness.
2
Tumor is 12.1–18 mm in diameter and ≤3 mm in thickness; or
Tumor is 9.1–15 mm in diameter and 3.1– 6 mm in thickness; or
Tumor is ≤12 mm in diameter and 6.1–9 mm in thickness.
3
Tumor is 15.1–18 mm in diameter and 3.1–6 mm in thickness; or
Tumor is 12.1–18 mm in diameter and 6.1–9 mm in thickness; or
Tumor is ≤18 mm in diameter and 9.1–12 mm in thickness; or
Tumor is ≤15 mm in diameter and 12.1–15 mm in thickness.
4
Tumor is >18 mm in diameter and may be any thickness; or
Tumor is 15.1–18 mm in diameter and >12 mm in thickness; or
Tumor is ≤15 mm in diameter and >15 mm in thickness.
Table 5. Definition of TNM Stage I Choroidal and Ciliary Body Melanomasa,b
Stage
TNM
Description
M = distant metastasis; N = regional lymph node; T = primary tumor.
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
b1) Primary ciliary body and choroidal melanomas are classified according to four tumor-size categories based on thickness and diameter. See Table 4. 2) In clinical practice, the largest tumor basal diameter may be estimated in optic disc diameters (DD) (average: 1 DD = 1.5 mm), and tumor thickness may be estimated in diopters (average: 2.5 diopters = 1 mm). Ultrasonography and fundus photography are used to provide more accurate measurements. 3) When histopathological measurements are recorded after fixation, tumor diameter and thickness may be underestimated because of tissue shrinkage.
I
T1a, N0, M0
–T1a = Tumor size category 1 without ciliary body involvement and extraocular extension.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
Table 6. Definition of TNM Stages IIA and IIB Choroidal and Ciliary Body Melanomasa,b
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis.
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
b1) Primary ciliary body and choroidal melanomas are classified according to four tumor-size categories based on thickness and diameter. See Table 4. 2) In clinical practice, the largest tumor basal diameter may be estimated in optic disc diameters (DD) (average: 1 DD = 1.5 mm), and tumor thickness may be estimated in diopters (average: 2.5 diopters = 1 mm). Ultrasonography and fundus photography are used to provide more accurate measurements. 3) When histopathological measurements are recorded after fixation, tumor diameter and thickness may be underestimated because of tissue shrinkage.
IIA
T1b–d, N0, M0
–T1b = Tumor size category 1 with ciliary body involvement.
–T1c = Tumor size category 1 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T1d = Tumor size category 1 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T2a, N0, M0
–T2a = Tumor size category 2 without ciliary body involvement and extraocular extension.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
IIB
T2b, N0, M0
–T2b = Tumor size category 2 with ciliary body involvement.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T3a, N0, M0
–T3a = Tumor size category 3 without ciliary body involvement and extraocular extension.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
Table 7. Definition of TNM Stages IIIA, IIIB, and IIIC Choroidal and Ciliary Body Melanomasa,b
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis.
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
b1) Primary ciliary body and choroidal melanomas are classified according to four tumor-size categories based on thickness and diameter. See Table 4. 2) In clinical practice, the largest tumor basal diameter may be estimated in optic disc diameters (DD) (average: 1 DD = 1.5 mm), and tumor thickness may be estimated in diopters (average: 2.5 diopters = 1 mm). Ultrasonography and fundus photography are used to provide more accurate measurements. 3) When histopathological measurements are recorded after fixation, tumor diameter and thickness may be underestimated because of tissue shrinkage.
IIIA
T2c–d, N0, M0
–T2c = Tumor size category 2 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T2d = Tumor size category 2 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T3b–c, N0, M0
–T3b = Tumor size category 3 with ciliary body involvement.
–T3c = Tumor size category 3 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T4a, N0, M0
–T4a = Tumor size category 4 without ciliary body involvement and extraocular extension.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
IIIB
T3d, N0, M0
–T3d = Tumor size category 3 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
T4b–c, N0, M0
–T4b = Tumor size category 4 with ciliary body involvement.
–T4c = Tumor size category 4 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
IIIC
T4d–e, N0, M0
–T4d = Tumor size category 4 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
–T4e = Any tumor size category with extraocular extension >5 mm in largest diameter.
N0 = No regional lymph node involvement.
M0 = No distant metastasis by clinical classification.
Table 8. Definition of TNM Stage IV Choroidal and Ciliary Body Melanomasa,b
Stage
TNM
Description
T = primary tumor; N = regional lymph node; M = distant metastasis.
aReprinted with permission from AJCC: Uveal melanoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 805–17.
b1) Primary ciliary body and choroidal melanomas are classified according to four tumor-size categories based on thickness and diameter. See Table 4. 2) In clinical practice, the largest tumor basal diameter may be estimated in optic disc diameters (DD) (average: 1 DD = 1.5 mm), and tumor thickness may be estimated in diopters (average: 2.5 diopters = 1 mm). Ultrasonography and fundus photography are used to provide more accurate measurements. 3) When histopathological measurements are recorded after fixation, tumor diameter and thickness may be underestimated because of tissue shrinkage.
IV
Any T, N1, M0
TX = Primary tumor cannot be assessed.
T0 = No evidence of primary tumor.
T1 = Tumor size category 1.
–T1a = Tumor size category 1 without ciliary body involvement and extraocular extension.
–T1b = Tumor size category 1 with ciliary body involvement.
–T1c = Tumor size category 1 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T1d = Tumor size category 1 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
T2 = Tumor size category 2.
–T2a = Tumor size category 2 without ciliary body involvement and extraocular extension.
–T2b = Tumor size category 2 with ciliary body involvement.
–T2c = Tumor size category 2 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T2d = Tumor size category 2 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
T3 = Tumor size category 3.
–T3a = Tumor size category 3 without ciliary body involvement and extraocular extension.
–T3b = Tumor size category 3 with ciliary body involvement.
–T3c = Tumor size category 3 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T3d = Tumor size category 3 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
T4 = Tumor size category 4.
–T4a = Tumor size category 4 without ciliary body involvement and extraocular extension.
–T4b = Tumor size category 4 with ciliary body involvement.
–T4c = Tumor size category 4 without ciliary body involvement but with extraocular extension ≤5 mm in largest diameter.
–T4d = Tumor size category 4 with ciliary body involvement and extraocular extension ≤5 mm in largest diameter.
–T4e = Any tumor size category with extraocular extension >5 mm in largest diameter.
N1 = Regional lymph node metastases or discrete tumor deposits in the orbit.
–N1a = Metastasis in one or more regional lymph nodes(s).
–N1b = No regional lymph nodes are positive, but there are discrete tumor deposits in the orbit that are not contiguous to the eye.
M0 = No distant metastasis by clinical classification.
Any T, Any N, M1a–c
Any T = See descriptions above in this table, stage IV, Any T, N1, M0.
NX = Regional lymph nodes cannot be assessed.
N0 = No regional lymph node involvement.
N1 = Regional lymph node metastases or discrete tumor deposits in the orbit.
–N1a = Metastasis in one or more regional lymph node(s).
–N1b = No regional lymph nodes are positive, but there are discrete tumor deposits in the orbit that are not contiguous to the eye (choroidal and ciliary body).
M1 = Distant metastasis.
–M1a = Largest diameter of the largest metastasis ≤3.0 cm.
–M1b = Largest diameter of the largest metastasis 3.1–8.0 cm.
–M1c = Largest diameter of the largest metastasis ≥8.1 cm.
Prognostic features
There are several key prognostic features that are important to collect in malignant melanoma of the uvea, even though they are not included in staging algorithms. These include:[7]
Molecular features
Chromosomal alterations.
Chromosome 3 status (loss or no loss, complete or partial).
Chromosome 6p status (gain or no gain).
Chromosome 8q status (gain or no gain).
Indicate:
Technique used for assessing chromosome status may include:
Karyotyping.
Fluorescence in situ hybridization.
Comparative genomic hybridization.
Loss of heterozygosity using DNA polymorphism analysis (e.g., single nucleotide polymorphism, microsatellite).
Other.
How specimen was obtained may include:
Enucleation.
Local resection.
Biopsy.
Fine-needle aspiration biopsy.
For needle biopsies, whether cytopathological evaluation was performed to confirm the presence of tumor cells.
Gene-expression profile: class 1 or class 2.
Indicate:
Technique used for gene-expression profiling may include:
Microarray.
Pathological complete response.
How specimen was obtained may include:
Enucleation.
Local resection.
Biopsy.
Fine-needle aspiration biopsy.
For needle biopsies, whether cytopathological evaluation was performed to confirm the presence of tumor cells.
Clinical and histopathological features
Clinical.
Positron emission tomography/computed tomography.
Fluorine F 18-fludeoxyglucose standardized uptake values (higher values in primary tumor may be associated with shorter survival).
Confocal indocyanine green angiography.
Identification of complex monocirculatory patterns (i.e., loops, networks, arcs with branching, parallel with cross-linking or a combination of these patterns may be associated with shorter survival).
Histopathological.
Mitotic count.
Number of mitotic figures per 40 high-power fields (typical field area 0.15–0.19 mm2, higher counts are associated with shorter survival).
Mean diameter of the ten largest nucleoli.
Mean of the longest nucleoli (MLN) is measured along a central 5-mm long strip, e.g., after silver staining (larger values are associated with shorter survival).
Presence of extravascular matrix patterns.
Loops.
Absent.
Present (shorter survival).
Loops forming networks.
Absent.
Present (shorter survival).
Other complex patterns (arcs with branching, parallel with cross-linking; absent or present).
The patterns are assessed with light microscopy under a dark green filter after staining with periodic-acid Schiff without counterstain.
Microvascular density.
Number of immunopositive elements labeled with markers for vascular endothelial cells (e.g., CD34 epitope, factor VIII-related antigen) in areas of densest vascularization (typical field area 0.31 mm2, higher counts are associated with shorter survival).
Insulin-like growth factor 1 receptor (IGF1-R).
Percentage of immunopositive tumor cells (high expression is associated with shorter survival).
Tumor-infiltrating lymphocytes.
Few (longest survival).
Moderate numbers.
Many (shortest survival).
Tumor-infiltrating macrophages.
Few (longest survival).
Moderate numbers.
Many (shortest survival).
The number can be compared with standard photographs.[12]
HLA class I expression.
Percentage of immunopositive tumor cells (low expression is associated with longer survival).
References
Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 115 (12): 1537-44, 1997. [PUBMED Abstract]
Diener-West M, Earle JD, Fine SL, et al.: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, II: characteristics of patients enrolled and not enrolled. COMS Report No. 17. Arch Ophthalmol 119 (7): 951-65, 2001. [PUBMED Abstract]
Shields CL, Shields JA, De Potter P, et al.: Diffuse choroidal melanoma. Clinical features predictive of metastasis. Arch Ophthalmol 114 (8): 956-63, 1996. [PUBMED Abstract]
Scott IU, Murray TG, Hughes JR: Evaluation of imaging techniques for detection of extraocular extension of choroidal melanoma. Arch Ophthalmol 116 (7): 897-9, 1998. [PUBMED Abstract]
Romero JM, Finger PT, Iezzi R, et al.: Three-dimensional ultrasonography of choroidal melanoma: extrascleral extension. Am J Ophthalmol 126 (6): 842-4, 1998. [PUBMED Abstract]
Echography (ultrasound) procedures for the Collaborative Ocular Melanoma Study (COMS), Report no. 12, Part I. J Ophthalmic Nurs Technol 18 (4): 143-9, 1999 Jul-Aug. [PUBMED Abstract]
Dithmar S, Diaz CE, Grossniklaus HE: Intraocular melanoma spread to regional lymph nodes: report of two cases. Retina 20 (1): 76-9, 2000. [PUBMED Abstract]
Diener-West M, Reynolds SM, Agugliaro DJ, et al.: Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol 123 (12): 1639-43, 2005. [PUBMED Abstract]
Shields CL, Santos MC, Shields JA, et al.: Extraocular extension of unrecognized choroidal melanoma simulating a primary optic nerve tumor: report of two cases. Ophthalmology 106 (7): 1349-52, 1999. [PUBMED Abstract]
Singh AD, Shields JA, Shields CL, et al.: Choroidal melanoma metastatic to the contralateral choroid. Am J Ophthalmol 132 (6): 941-3, 2001. [PUBMED Abstract]
Mäkitie T, Summanen P, Tarkkanen A, et al.: Tumor-infiltrating macrophages (CD68(+) cells) and prognosis in malignant uveal melanoma. Invest Ophthalmol Vis Sci 42 (7): 1414-21, 2001. [PUBMED Abstract]
Treatment Option Overview for Intraocular (Uveal) Melanoma
Observation
Patients with iris melanomas have relatively good outcomes, with 5-year survival rates exceeding 95%. Iris melanomas are predominantly of the spindle-cell type and are usually smaller in size than posterior melanomas because they are detected earlier. Conservative management is generally advocated whenever possible, but surgical intervention may be justified with unequivocal tumor growth or with extensive disease at initial examination.
The management of small choroidal melanomas is controversial, and it is not clear whether treating small tumors prevents metastasis.[1] The natural history of small choroidal melanomas is poorly understood. Small, pigmented, choroidal lesions cannot always be differentiated reliably on examination. Growth is a presumed indicator of malignant potential.[2] The likelihood of progression from the time of diagnosis to the time when tumor growth warrants treatment has not been well characterized. Some ophthalmologists advocate observation. This course has been justified on several grounds, including the difficulty of establishing a correct diagnosis, the lack of any documented efficacy for globe-conserving treatments, and concerns for severe treatment-related morbidity. Others have advocated earlier therapeutic intervention.[1,3,4]
Although patients diagnosed with small choroidal tumors were not eligible for participation in the Collaborative Ocular Melanoma Study (COMS), these patients could participate in a prospective follow-up study to evaluate the natural history of small lesions. The 2-year tumor growth estimates were 21%, and the 5-year tumor growth estimates were 31%.[5] Clinical risk factors associated with tumor growth included:[3,5]
Increased tumor thickness.
Presence of subretinal fluid.
Orange pigmentation.
Absence of drusen.
Absence of retinal pigment.
Margin at the optic disc.
Epithelial changes surrounding the tumor.
Surgery
The selection of treatment depends on the following:
Site of origin (choroid, ciliary body, or iris).
Size and location of the lesion.
Age of the patient.
Occurrence of extraocular invasion, recurrence, or metastasis.
Enucleation
In the past, enucleation (eye removal) was the standard treatment for primary choroidal melanoma, and it is still used when large tumors are present. However, enucleation has been largely replaced by radiation therapy (i.e., brachytherapy with radioactive plaques or external-beam, charged-particle radiation therapy) to spare the affected eye.[6,7]
A randomized trial evaluated the role of pre-enucleation EBRT for large choroidal tumors that required enucleation. The study found that EBRT had no impact on overall survival (OS).[8,9][Level of evidence A1] A COMS trial included 1,003 patients with large choroidal melanomas (≥2 mm in height and ≥16 mm in diameter, or ≥10 mm in height irrespective of diameter, or ≥8 mm in height and border <2 mm from the optic disc) with no known metastases. Patients were randomly assigned to receive enucleation alone or enucleation after preoperative external photon-beam radiation from cobalt 60 or accelerators (20 Gy in 5 daily fractions) to the orbit and globe.[8,9] Through 10 years of follow-up, the median survival in both arms was approximately 7 years, and the 10-year all-cause mortality rate was 61% in both arms (relative risk [RR]death, 1.00; 95% confidence interval [CI], 0.85–1.18). Metastasis-free survival was also nearly identical in both arms.
Transscleral local resection
Eye-sparing transscleral local resection plays a limited role in the management of uveal melanoma. It is used in patients with large choroidal and ciliary body tumors who are not candidates for radiation therapy but are highly motivated to retain their eye.[10–12] The procedure is technically demanding and is generally performed only in centers with specialized expertise in this surgery. There is a substantial risk of retinal detachment, intraocular bleeding, and complications associated with the anesthesia-induced hypotension used to decrease the risk of bleeding. Either adjuvant brachytherapy or neoadjuvant proton-beam therapy are administered. Experience is limited to retrospective, single-center, case series.[10–12][Level of evidence C3]
Surgical resection of metastases
Surgical resection of metastases from ocular melanoma has been reported in case series of highly selected patients with occasional favorable outcomes.[13,14] However, the favorable outcomes may be the result of strong patient-selection factors, and the role of resection in this setting is unclear.[13,14][Level of evidence C3]
Radiation Therapy
Episcleral brachytherapy using plaques containing small radioactive seeds is the most common form of radiation used in the management of intraocular melanoma. Iodine I 125 (125I), cobalt Co 60 (60Co), palladium Pd 103 (103Pd), iridium Ir 192 (192Ir), and ruthenium Ru 106 (106Ru) are examples of radioactive isotopes used in the brachytherapy plaques. Isotopes with relatively low photon and electron emissions (125I, 103Pd, and 106Ru) are more easily shielded to reduce the exposure to adjacent normal tissues, and 125I is probably the most commonly used radioisotope.[15] Although plaque radiation therapy allows preservation of the eye, visual acuity is frequently lost over time.
In a case series of 1,106 patients who were treated with plaque radiation therapy for uveal melanoma and who had an initial acuity of at least 20/100, 68% developed poor acuity (i.e., 20/200 or worse) within 10 years.[16]
Factors associated with worse acuity outcomes included:[16]
Age older than 60 years.
Diminished baseline acuity.
Diabetes.
Increased tumor size and thickness.
Location near the fovea or optic disc.
Type of isotope therapy (106Ru, 60Co, or 192Ir vs. 125I).
125I brachytherapy yields equivalent overall and melanoma metastasis-specific survival rates to enucleation for medium-sized melanomas.[17][Level of evidence A1] The randomized COMS Medium Tumor Trial compared 125I episcleral-plaque brachytherapy (85 Gy at 0.42–1.06 Gy/hr) with enucleation in 1,317 patients with medium-sized choroidal tumors (tumor height 2.5–10.0 mm and tumor diameter ≤16.0 mm that were not contiguous with the optic disc).[17] Eighty-five percent of the patients treated with 125I brachytherapy retained their eye for 5 years or more, and 37% of them had visual acuity better than 20/200 in the irradiated eye 5 years after treatment.[17] No statistically significant differences in mortality were observed between the two study arms after 12 years of follow-up, either for death from all causes or death with histopathologically confirmed melanoma metastasis.[18] The 5- and 10-year all-cause mortality rates were 19% and 35% in both study arms. The cumulative all-cause mortality rate at 12 years was 43% in the 125I arm versus 41% in the enucleation arm (RR, 1.04; 95% CI, 0.86–1.24). The 5-year metastasis-specific mortality rates were 13% in both arms; at 10 years, the rates were 21% and 22% (RR for metastasis-specific mortality, 1.07; 95% CI, 0.81–1.41 through 12 years).
In a companion study within the COMS, 209 patients were prospectively assessed for quality of life during the first 5 years of follow-up.[19] Both study groups reported increasing difficulty with vision-oriented daily activities and ocular pain as time elapsed. Most measures of visual function were similar between the two groups. However, there were statistically significant differences favoring the brachytherapy group in comfort with driving for the first year after therapy and in reported peripheral vision for the first 2 years after therapy. These differences disappeared by year 5 of follow-up.[19][Level of evidence A3]
Charged-particle EBRT (using protons, carbon ions, or helium ions) is the other major form of radiation therapy used in the management of ocular melanomas.[20–23] This form of radiation therapy requires sophisticated equipment available only at selected centers. In addition, charged-particle EBRT involves patient cooperation during treatment (e.g., voluntarily fixating the eye on a particular point so the tumor is positioned properly in the radiation beam). A lower risk of early and late local radiation failures has been reported after charged-particle EBRT than after the use of brachytherapy, possibly resulting from differences in dose distribution in the two techniques.[20][Levels of evidence B3 and C3]
In a single-center, single-surgeon study, 184 patients with uveal melanomas smaller than 15 mm in diameter and smaller than 10 mm in thickness were randomly assigned to receive 125I brachytherapy versus helium ion radiation (to an estimated dose of 70 Gy equivalents in 5 fractions over 7 to 11 days in each arm).[24] The local tumor regrowth rate by 4 years was 13.3% in the brachytherapy arm compared with 0% in the helium ion arm (P < .001). However, the rates of metastasis, death from metastasis, and overall mortality were similar in both arms.[24][Level of evidence B3]
Because of its dose distribution, charged-particle irradiation can be better used than plaque brachytherapy to treat larger tumors and tumors closer to the fovea or optic disc. A large, single-center, single-surgeon series of 2,069 patients treated with proton-beam therapy had an actuarial local control rate of 95% (95% CI, 93%–96%) at 15 years. The cumulative rate of enucleation was 16% (95% CI, 13%–20%). Enucleations were primarily the result of neovascular glaucoma (46% of enucleations), blind uncomfortable eyes (31% of enucleations), or local recurrence (23% of enucleations). As with plaque radiation, risk factors for deterioration in visual acuity after charged-particle radiation were tumor size, location near the fovea or optic disc, baseline acuity, and underlying diabetes.[21]
Similarly, another large, single-center, single-surgeon, consecutive series of 886 patients treated with proton-beam irradiation reported a local control rate of 92.1% (95% CI, 89.8%–94.6%) and ocular conservation rate of 87.3% (95% CI, 83.9%–90.9%) at 10 years.[22][Level of evidence C3] The actuarial OS rate at 10 years was 64.1% (95% CI, 59.5%–69.0%).
In a single-center, phase I/II study of 57 evaluable patients treated with carbon ion-beam irradiation and followed for a median of 26 months, 26 patients developed neovascular glaucoma or severe eye pain from increased intraocular pressure, and 3 patients underwent enucleation. One patient had a local tumor recurrence.[23]
In an attempt to lower the complication rate and improve functional outcomes, one study compared a decreased dose of 50 cobalt Gy equivalents (CGE) with 70 CGE proton beam (each delivered in 5 fractions, usually within a 7-day period). Patients (n = 188) with tumors smaller than 15 mm in diameter and smaller than 5 mm in height, which were located near the optic disc or macula, were randomly assigned to the two doses in a double-masked study design. At 5 years, there were no statistically significant differences in local tumor control, rate of metastasis, visual acuity, or complication rates. However, the visual fields were better in the 50-CGE group.[25][Level of evidence B3]
As noted in the Surgery section, the role of pre-enucleation external photon-beam radiation therapy has been tested in a randomized trial and has shown no impact on OS for large choroidal tumors treated with enucleation.[8,9]
External-beam–photon-beam (gamma-ray) radiation therapy with gamma-knife stereotactic radiation surgery as a single-fraction [26] or fractionated stereotactic radiation [27,28] is being investigated as an alternative to brachytherapy or charged-beam radiation for posterior uveal melanomas, particularly for tumors too large or too close to the optic disc or macula to treat with brachytherapy. Because the dose rate of radiation has a slower delivery time than is the case with charged particles, specialized techniques are used to immobilize the eye [26] or to avoid delivery of the photons while the eye is moving or closed.[28] Experience is more limited with external-beam–photon therapy than for either brachytherapy or charged-particle EBRT, and there are no controlled comparisons with either of the other techniques. Early results from single-center series suggest similar levels of local tumor control and eye retention rates, but patient-selection factors may play a role.[28][Level of evidence C3]
Transpupillary Thermotherapy
Transpupillary thermotherapy (TTT) directs an infrared laser, usually at a wavelength of 810 nm, through a dilated pupil in one or more sessions to induce heat necrosis of uveal melanomas. This method carries the theoretical advantage of high-precision destruction of tumor tissue under direct visualization. However, TTT has important limitations that confine its use to specific circumstances.[1,29] The limited ability of TTT to penetrate thick tumors with sufficient energy restricts its use to small melanomas or tumors of a size that some ophthalmologists recommend for follow-up without any initial therapy. For more information, see the Observation section. When used as the primary therapy, there are relatively high rates of local recurrence and retinal vascular damage. Recurrence rates are particularly high when the tumor abuts the optic nerve and overhangs the optic disc.[1][Level of evidence C3]
In a single-center study, 95 patients with small choroidal melanomas (diameter <10 mm and thickness <3.5 mm) were randomly assigned to receive either TTT or 125I brachytherapy (100 Gy).[30] The tumor regression rate was 92% in the TTT arm and 98% in the 125I arm (P = .4). With a mean follow-up time of 56.2 months, there were four recurrences in the TTT arm and one in the 125I arm. However, the study was too small to provide clear information on efficacy differences.
TTT is also under evaluation as an adjunct to primary therapy with proton-beam radiation. In large uveal melanomas, proton-beam therapy is associated with exudative, inflammatory, and glaucomatous complications that may require enucleation. In a single-center trial, 151 patients with uveal melanomas at least 7 mm thick or at least 15 mm in diameter were randomly assigned to receive proton-beam radiation (60 CGEs over four daily fractions) with or without TTT (810 nm wavelength at 1, 6, and 12 months after therapy) and followed for a median of 38 months.[31] There were no differences between the two groups in maculopathy, papillopathy, or glaucoma. The enucleation rate was lower in the TTT group (about 2% vs. 18% at 5 years, P = .02). However, the study was not masked, and replication of the results would be important.
There are uncertainties at all stages about the optimal management of intraocular melanoma. Physicians should discuss clinical trial opportunities with eligible patients. Information about ongoing clinical trials is available from the NCI website.
References
Shields CL, Shields JA, Perez N, et al.: Primary transpupillary thermotherapy for small choroidal melanoma in 256 consecutive cases: outcomes and limitations. Ophthalmology 109 (2): 225-34, 2002. [PUBMED Abstract]
Augsburger JJ: Is observation really appropriate for small choroidal melanomas. Trans Am Ophthalmol Soc 91: 147-68; discussion 169-75, 1993. [PUBMED Abstract]
Shields CL, Cater J, Shields JA, et al.: Combination of clinical factors predictive of growth of small choroidal melanocytic tumors. Arch Ophthalmol 118 (3): 360-4, 2000. [PUBMED Abstract]
Robertson DM, Buettner H, Bennett SR: Transpupillary thermotherapy as primary treatment for small choroidal melanomas. Arch Ophthalmol 117 (11): 1512-9, 1999. [PUBMED Abstract]
Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 115 (12): 1537-44, 1997. [PUBMED Abstract]
Zimmerman LE, McLean IW, Foster WD: Statistical analysis of follow-up data concerning uveal melanomas, and the influence of enucleation. Ophthalmology 87 (6): 557-64, 1980. [PUBMED Abstract]
De Potter P, Shields CL, Shields JA: New treatment modalities for uveal melanoma. Curr Opin Ophthalmol 7 (3): 27-32, 1996. [PUBMED Abstract]
The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma II: initial mortality findings. COMS report no. 10. Am J Ophthalmol 125 (6): 779-96, 1998. [PUBMED Abstract]
Hawkins BS; Collaborative Ocular Melanoma Study Group: The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma: IV. Ten-year mortality findings and prognostic factors. COMS report number 24. Am J Ophthalmol 138 (6): 936-51, 2004. [PUBMED Abstract]
Damato B: The role of eyewall resection in uveal melanoma management. Int Ophthalmol Clin 46 (1): 81-93, 2006. [PUBMED Abstract]
Bechrakis NE, Bornfeld N, Zöller I, et al.: Iodine 125 plaque brachytherapy versus transscleral tumor resection in the treatment of large uveal melanomas. Ophthalmology 109 (10): 1855-61, 2002. [PUBMED Abstract]
Bechrakis NE, Petousis V, Willerding G, et al.: Ten-year results of transscleral resection of large uveal melanomas: local tumour control and metastatic rate. Br J Ophthalmol 94 (4): 460-6, 2010. [PUBMED Abstract]
Hsueh EC, Essner R, Foshag LJ, et al.: Prolonged survival after complete resection of metastases from intraocular melanoma. Cancer 100 (1): 122-9, 2004. [PUBMED Abstract]
Pawlik TM, Zorzi D, Abdalla EK, et al.: Hepatic resection for metastatic melanoma: distinct patterns of recurrence and prognosis for ocular versus cutaneous disease. Ann Surg Oncol 13 (5): 712-20, 2006. [PUBMED Abstract]
Albert DM, Kulkarni AD: Intraocular melanoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 2090-8.
Shields CL, Shields JA, Cater J, et al.: Plaque radiotherapy for uveal melanoma: long-term visual outcome in 1106 consecutive patients. Arch Ophthalmol 118 (9): 1219-28, 2000. [PUBMED Abstract]
Diener-West M, Earle JD, Fine SL, et al.: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, III: initial mortality findings. COMS Report No. 18. Arch Ophthalmol 119 (7): 969-82, 2001. [PUBMED Abstract]
Collaborative Ocular Melanoma Study Group: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report No. 28. Arch Ophthalmol 124 (12): 1684-93, 2006. [PUBMED Abstract]
Melia M, Moy CS, Reynolds SM, et al.: Quality of life after iodine 125 brachytherapy vs enucleation for choroidal melanoma: 5-year results from the Collaborative Ocular Melanoma Study: COMS QOLS Report No. 3. Arch Ophthalmol 124 (2): 226-38, 2006. [PUBMED Abstract]
Char DH, Kroll S, Phillips TL, et al.: Late radiation failures after iodine 125 brachytherapy for uveal melanoma compared with charged-particle (proton or helium ion) therapy. Ophthalmology 109 (10): 1850-4, 2002. [PUBMED Abstract]
Gragoudas E, Li W, Goitein M, et al.: Evidence-based estimates of outcome in patients irradiated for intraocular melanoma. Arch Ophthalmol 120 (12): 1665-71, 2002. [PUBMED Abstract]
Caujolle JP, Mammar H, Chamorey E, et al.: Proton beam radiotherapy for uveal melanomas at nice teaching hospital: 16 years’ experience. Int J Radiat Oncol Biol Phys 78 (1): 98-103, 2010. [PUBMED Abstract]
Tsuji H, Ishikawa H, Yanagi T, et al.: Carbon-ion radiotherapy for locally advanced or unfavorably located choroidal melanoma: a Phase I/II dose-escalation study. Int J Radiat Oncol Biol Phys 67 (3): 857-62, 2007. [PUBMED Abstract]
Char DH, Quivey JM, Castro JR, et al.: Helium ions versus iodine 125 brachytherapy in the management of uveal melanoma. A prospective, randomized, dynamically balanced trial. Ophthalmology 100 (10): 1547-54, 1993. [PUBMED Abstract]
Gragoudas ES, Lane AM, Regan S, et al.: A randomized controlled trial of varying radiation doses in the treatment of choroidal melanoma. Arch Ophthalmol 118 (6): 773-8, 2000. [PUBMED Abstract]
Modorati G, Miserocchi E, Galli L, et al.: Gamma knife radiosurgery for uveal melanoma: 12 years of experience. Br J Ophthalmol 93 (1): 40-4, 2009. [PUBMED Abstract]
Muller K, Nowak PJ, de Pan C, et al.: Effectiveness of fractionated stereotactic radiotherapy for uveal melanoma. Int J Radiat Oncol Biol Phys 63 (1): 116-22, 2005. [PUBMED Abstract]
Dieckmann K, Georg D, Bogner J, et al.: Optimizing LINAC-based stereotactic radiotherapy of uveal melanomas: 7 years’ clinical experience. Int J Radiat Oncol Biol Phys 66 (4 Suppl 1): 47-52, 2006.
Harbour JW, Meredith TA, Thompson PA, et al.: Transpupillary thermotherapy versus plaque radiotherapy for suspected choroidal melanomas. Ophthalmology 110 (11): 2207-14; discussion 2215, 2003. [PUBMED Abstract]
Pilotto E, Vujosevic S, De Belvis V, et al.: Long-term choroidal vascular changes after iodine brachytherapy versus transpupillary thermotherapy for choroidal melanoma. Eur J Ophthalmol 19 (4): 646-53, 2009 Jul-Aug. [PUBMED Abstract]
Desjardins L, Lumbroso-Le Rouic L, Levy-Gabriel C, et al.: Combined proton beam radiotherapy and transpupillary thermotherapy for large uveal melanomas: a randomized study of 151 patients. Ophthalmic Res 38 (5): 255-60, 2006. [PUBMED Abstract]
Treatment of Iris Melanoma
Melanocytic stromal proliferations and nevi of the iris are the most common tumors of the iris, but melanoma is rare.[1,2] Clinical differentiation between an iris nevus and a melanoma might sometimes be difficult and, at times, impossible. Melanomas of the iris are usually small discrete lesions, although they may occasionally be diffuse, infiltrative, or multiple and may result in heterochromia, chronic uveitis, or spontaneous hemorrhage into the anterior chamber of the eye (hyphema). Iris melanomas that involve more than 66% of the angle circumference are associated with secondary glaucoma.[3]
Routine evaluation of iris melanomas includes gonioscopy, transillumination of the globe, and indirect ophthalmoscopy with 360° of scleral depression. Photographic documentation is essential to verify progression in size or growth of the tumor.[4] Anterior segment fluorescein angiography may be helpful to demonstrate the vascularity of the lesion but is not diagnostic. High-resolution ultrasound biomicroscopy can be used to measure small lesions (basal dimensions and thickness) and to assess tumor involvement of the anterior ciliary body, angle, and overlying sclera.[5] The main disadvantage with this technology is its limited penetration of large lesions. In these cases, conventional ultrasonography is more accurate.
In general, patients with iris melanomas have relatively good outcomes. Only about 3% of these melanomas metastasize within 5 years.[1] Iris melanomas are predominantly of the spindle-cell type and are usually smaller in size than posterior melanomas. Clinical features, including prominent tumor vascularity, rapid growth, and heterogeneous pigmentation, are associated with an epithelioid cell component.[6] Involvement of the iridocorneal angles is frequently associated with ciliary body invasion.[6]
Because iris melanomas are rare and have a good prognosis, clinical trials with sufficient power are impractical. Therefore, treatment experience is based principally on case series and case reports. Conservative management is generally advocated whenever possible, but surgical intervention may be justified with unequivocal tumor growth or extensive disease at initial examination.
Treatment Options for Iris Melanoma
Treatment options for iris melanoma include:
Observation with careful follow-up. This option is used in asymptomatic patients with stable lesions; follow-up includes serial photography.[3]
Local resection. This option is used when progressive and pronounced growth is documented.[2]
Enucleation. This option is used if the tumor is not amenable to local resection because of diffuse involvement of the iris, involvement of more than 50% of the iris and anterior chamber angle, intractable glaucoma, or extraocular extension.[7]
Plaque radiation therapy. This option is offered as an alternative for large, diffuse, surgically nonresectable lesions of the iris.[8]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Albert DM, Kulkarni AD: Intraocular melanoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 2090-8.
Starr OD, Patel DV, Allen JP, et al.: Iris melanoma: pathology, prognosis and surgical intervention. Clin Experiment Ophthalmol 32 (3): 294-6, 2004. [PUBMED Abstract]
Marcus DM, Sahel JA, Jakobiec FA, et al.: Pigmented tumors of the iris. In: Albert DM, Jakobiec FA, eds.: Principles and Practice of Ophthalmology. WB Saunders Co., 1994, pp 3198-3208.
Conway RM, Chua WC, Qureshi C, et al.: Primary iris melanoma: diagnostic features and outcome of conservative surgical treatment. Br J Ophthalmol 85 (7): 848-54, 2001. [PUBMED Abstract]
Melanocytic tumors of the iris stroma. In: Shields JA: Diagnosis and Management of Intraocular Tumors. C.V. Mosby Company, 1983, pp 83-121.
Shields CL, Shields JA, De Potter P, et al.: Treatment of non-resectable malignant iris tumours with custom designed plaque radiotherapy. Br J Ophthalmol 79 (4): 306-12, 1995. [PUBMED Abstract]
Treatment of Ciliary Body Melanoma
Melanoma involving the ciliary body is rare and patients have a poor prognosis. In some cases, diagnosis may be difficult because of similarity to other eye diseases. The differential diagnosis of ciliary body melanoma is considered in cases of unilateral pigmentary glaucoma and chronic uveitis.[1]
Ultrasound biomicroscopy can be used to evaluate tumor shape, thickness, margins, reflectivity, and local invasion.[2,3] Patients with tumors greater than 7 mm in thickness are at increased risk of metastatic disease and melanoma-related death compared with patients with thinner tumors.[4]
Treatment Options for Ciliary Body Melanoma
There are several options for management of ciliary body melanoma. All of them are reported from case series.[Level of evidence C3] The choice of therapy, however, depends on many factors.
Treatment options for ciliary body melanoma include:
Plaque radiation therapy. Local control rates are high, but treatment is associated with a high incidence of secondary cataract.[4,5]
External-beam, charged-particle radiation therapy. This approach is offered at specialized referral centers. It requires careful patient cooperation, with voluntary fixation of gaze.[6–8]
Local tumor resection. This option is mainly suitable for selected ciliary body or anterior choroidal tumors with smaller basal dimension and greater thickness.[9,10]
Enucleation. This option is generally reserved for large melanomas when regaining useful vision is unlikely. It is also indicated in the presence of intractable secondary glaucoma and extraocular extension.[5,8]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Nguyen QD, Foster CS: Ciliary body melanoma masquerading as chronic uveitis. Ocul Immunol Inflamm 6 (4): 253-6, 1998. [PUBMED Abstract]
Marigo FA, Finger PT, McCormick SA, et al.: Iris and ciliary body melanomas: ultrasound biomicroscopy with histopathologic correlation. Arch Ophthalmol 118 (11): 1515-21, 2000. [PUBMED Abstract]
Daftari I, Barash D, Lin S, et al.: Use of high-frequency ultrasound imaging to improve delineation of anterior uveal melanoma for proton irradiation. Phys Med Biol 46 (2): 579-90, 2001. [PUBMED Abstract]
Gündüz K, Shields CL, Shields JA, et al.: Plaque radiotherapy of uveal melanoma with predominant ciliary body involvement. Arch Ophthalmol 117 (2): 170-7, 1999. [PUBMED Abstract]
Finger PT: Plaque radiation therapy for malignant melanoma of the iris and ciliary body. Am J Ophthalmol 132 (3): 328-35, 2001. [PUBMED Abstract]
Munzenrider JE: Uveal melanomas. Conservation treatment. Hematol Oncol Clin North Am 15 (2): 389-402, 2001. [PUBMED Abstract]
Char DH, Kroll SM, Castro J: Ten-year follow-up of helium ion therapy for uveal melanoma. Am J Ophthalmol 125 (1): 81-9, 1998. [PUBMED Abstract]
De Potter P: [Choroidal melanoma: current therapeutic approaches] J Fr Ophtalmol 25 (2): 203-11, 2002. [PUBMED Abstract]
De Potter P, Shields CL, Shields JA: New treatment modalities for uveal melanoma. Curr Opin Ophthalmol 7 (3): 27-32, 1996. [PUBMED Abstract]
A wide range of 5-year mortality rates has been reported for patients with small choroidal melanomas, with an average rate of about 16%.[1,2] Several studies indicate that the two most important clinical factors predictive of mortality are larger tumor size at the time of treatment and documentation of tumor growth.[3]
The management of small choroidal melanomas is controversial. The likelihood of progression from the time of diagnosis to growth warranting treatment has not been well characterized. Many ophthalmologists advocate initial observation. This initial management strategy is justified on several grounds, including difficulty in establishing a correct diagnosis, lack of documented efficacy for globe-conserving treatments, and concerns for severe treatment-related morbidity. Others have advocated earlier therapeutic intervention.[4–6]
Treatment Options for Small Choroidal Melanoma
Treatment options for small choroidal melanoma include:
Observation. This strategy is important for patients with an uncertain diagnosis or whose tumor growth has not been documented. It is also used for asymptomatic patients with stable lesions (particularly older or debilitated patients) and for patients with a tumor in their only useful eye.[2]
Plaque radiation therapy. This treatment is used for small- or medium-sized uveal melanomas, amelanotic tumors, or tumors that touch the optic disc for greater than 3 clock-hours of optic disk circumference.[7,8]
External-beam, charged-particle radiation therapy. This approach is offered at specialized referral centers. It requires careful patient cooperation, with voluntary fixation of gaze.[7–10]
Gamma-knife radiation surgery. This treatment may be a feasible option for small- to medium-sized melanomas.[11–13]
Transpupillary thermotherapy. As noted above, this approach has very limited use, but it can be used as a primary treatment or as an adjunctive method to plaque radiation therapy.[5,6,14–18] For more information, see the Transpupillary Thermotherapy section.
Local tumor resection. This strategy is used mainly for selected ciliary body or anterior choroidal tumors with smaller basal dimensions and greater thickness.[19]
Enucleation. This approach is used when severe intraocular pressure elevation is a factor. It may also be considered with small- and medium-sized melanomas that are invading the tissues of the optic nerve.[20]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Diener-West M, Hawkins BS, Markowitz JA, et al.: A review of mortality from choroidal melanoma. II. A meta-analysis of 5-year mortality rates following enucleation, 1966 through 1988. Arch Ophthalmol 110 (2): 245-50, 1992. [PUBMED Abstract]
Mortality in patients with small choroidal melanoma. COMS report no. 4. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 115 (7): 886-93, 1997. [PUBMED Abstract]
Shields CL, Shields JA, Kiratli H, et al.: Risk factors for growth and metastasis of small choroidal melanocytic lesions. Ophthalmology 102 (9): 1351-61, 1995. [PUBMED Abstract]
Shields CL, Cater J, Shields JA, et al.: Combination of clinical factors predictive of growth of small choroidal melanocytic tumors. Arch Ophthalmol 118 (3): 360-4, 2000. [PUBMED Abstract]
Shields CL, Shields JA, Perez N, et al.: Primary transpupillary thermotherapy for small choroidal melanoma in 256 consecutive cases: outcomes and limitations. Ophthalmology 109 (2): 225-34, 2002. [PUBMED Abstract]
Robertson DM, Buettner H, Bennett SR: Transpupillary thermotherapy as primary treatment for small choroidal melanomas. Arch Ophthalmol 117 (11): 1512-9, 1999. [PUBMED Abstract]
Munzenrider JE: Uveal melanomas. Conservation treatment. Hematol Oncol Clin North Am 15 (2): 389-402, 2001. [PUBMED Abstract]
Char DH, Kroll SM, Castro J: Ten-year follow-up of helium ion therapy for uveal melanoma. Am J Ophthalmol 125 (1): 81-9, 1998. [PUBMED Abstract]
Woodburn R, Danis R, Timmerman R, et al.: Preliminary experience in the treatment of choroidal melanoma with gamma knife radiosurgery. J Neurosurg 93 (Suppl 3): 177-9, 2000. [PUBMED Abstract]
Modorati G, Miserocchi E, Galli L, et al.: Gamma knife radiosurgery for uveal melanoma: 12 years of experience. Br J Ophthalmol 93 (1): 40-4, 2009. [PUBMED Abstract]
Muller K, Nowak PJ, de Pan C, et al.: Effectiveness of fractionated stereotactic radiotherapy for uveal melanoma. Int J Radiat Oncol Biol Phys 63 (1): 116-22, 2005. [PUBMED Abstract]
Shields JA, Shields CL: Atlas of Intraocular Tumors. Lippincott Williams & Wilkins, 1999.
Treatment of Medium and Large Choroidal Melanoma
Eye-sparing radiation therapy, either by plaque brachytherapy or external beam, is the preferred option for most patients with medium-sized choroidal melanoma. Enucleation remains the standard therapy for large choroidal melanomas and melanomas that cause severe glaucoma or invade the optic nerve.
Treatment Options for Medium and Large Choroidal Melanoma
Tumor growth pattern is a factor in the therapeutic decision. Enucleation is considered for a diffuse melanoma or extraocular extension, but radiation therapy can be employed for less extensive disease.
Treatment options for medium and large choroidal melanoma include:
External-beam, charged-particle radiation therapy. This approach is offered at specialized referral centers. It requires careful patient cooperation, with voluntary fixation of gaze.[5–7]
Combined therapy, with ablative laser coagulation or transpupillary thermotherapy to supplement plaque treatment.[10,11] For more information, see the Transpupillary Thermotherapy section.
Enucleation. This approach is considered primarily for diffuse melanomas or extraocular extension. Enucleation may be necessary when there are radiation complications or tumor recurrences.[10]
Large choroidal melanomas
Enucleation when the tumor is judged to be too large for eye-sparing approaches.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Karvat A, Duzenli C, Ma R, et al.: The treatment of choroidal melanoma with 198 Au plaque brachytherapy. Radiother Oncol 59 (2): 153-6, 2001. [PUBMED Abstract]
Tabandeh H, Chaudhry NA, Murray TG, et al.: Intraoperative echographic localization of iodine-125 episcleral plaque for brachytherapy of choroidal melanoma. Am J Ophthalmol 129 (2): 199-204, 2000. [PUBMED Abstract]
Diener-West M, Earle JD, Fine SL, et al.: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, III: initial mortality findings. COMS Report No. 18. Arch Ophthalmol 119 (7): 969-82, 2001. [PUBMED Abstract]
Melia BM, Abramson DH, Albert DM, et al.: Collaborative ocular melanoma study (COMS) randomized trial of I-125 brachytherapy for medium choroidal melanoma. I. Visual acuity after 3 years COMS report no. 16. Ophthalmology 108 (2): 348-66, 2001. [PUBMED Abstract]
Char DH, Quivey JM, Castro JR, et al.: Helium ions versus iodine 125 brachytherapy in the management of uveal melanoma. A prospective, randomized, dynamically balanced trial. Ophthalmology 100 (10): 1547-54, 1993. [PUBMED Abstract]
Fuss M, Loredo LN, Blacharski PA, et al.: Proton radiation therapy for medium and large choroidal melanoma: preservation of the eye and its functionality. Int J Radiat Oncol Biol Phys 49 (4): 1053-9, 2001. [PUBMED Abstract]
Char DH, Kroll SM, Castro J: Ten-year follow-up of helium ion therapy for uveal melanoma. Am J Ophthalmol 125 (1): 81-9, 1998. [PUBMED Abstract]
Peyman GA, Juarez CP, Diamond JG, et al.: Ten years experience with eye wall resection for uveal malignant melanomas. Ophthalmology 91 (12): 1720-5, 1984. [PUBMED Abstract]
Seregard S, Landau I: Transpupillary thermotherapy as an adjunct to ruthenium plaque radiotherapy for choroidal melanoma. Acta Ophthalmol Scand 79 (1): 19-22, 2001. [PUBMED Abstract]
Shields JA: The expanding role of laser photocoagulation for intraocular tumors. The 1993 H. Christian Zweng Memorial Lecture. Retina 14 (4): 310-22, 1994. [PUBMED Abstract]
Treatment of Extraocular Extension and Metastatic Intraocular Melanoma
Extrascleral extension is associated with a poor prognosis. For patients with gross tumor involvement of the orbit, treatment requires orbital exenteration. However, there is no evidence that such radical surgery will prolong survival. Most patients with localized or encapsulated extraocular extension are not exenterated. This subject is controversial.[1–5]
No effective method of systemic treatment has been identified for patients with metastatic ocular melanoma. Clinical trials are an option for these patients.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Shammas HF, Blodi FC: Prognostic factors in choroidal and ciliary body melanomas. Arch Ophthalmol 95 (1): 63-9, 1977. [PUBMED Abstract]
Pach JM, Robertson DM, Taney BS, et al.: Prognostic factors in choroidal and ciliary body melanomas with extrascleral extension. Am J Ophthalmol 101 (3): 325-31, 1986. [PUBMED Abstract]
Kersten RC, Tse DT, Anderson RL, et al.: The role of orbital exenteration in choroidal melanoma with extrascleral extension. Ophthalmology 92 (3): 436-43, 1985. [PUBMED Abstract]
Hykin PG, McCartney AC, Plowman PN, et al.: Postenucleation orbital radiotherapy for the treatment of malignant melanoma of the choroid with extrascleral extension. Br J Ophthalmol 74 (1): 36-9, 1990. [PUBMED Abstract]
Gündüz K, Shields CL, Shields JA, et al.: Plaque radiotherapy for management of ciliary body and choroidal melanoma with extraocular extension. Am J Ophthalmol 130 (1): 97-102, 2000. [PUBMED Abstract]
Treatment of Recurrent Intraocular Melanoma
The prognosis for any patient with recurring or relapsing disease is poor, regardless of cell type or stage. The question and selection of further treatment depends on many factors, including the extent of the lesion, age and health of the patient, prior treatment, site of recurrence, and individual patient considerations. Surgical resection of metastases diagnosed after initial management of ocular melanoma in single-center case series of highly selected patients has been reported. The extent to which the occasional favorable outcomes are the result of strong selection factors is not clear, so this approach cannot be considered standard.[1]
Eligible patients should consider participation in clinical trials whenever possible.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Hsueh EC, Essner R, Foshag LJ, et al.: Prolonged survival after complete resection of metastases from intraocular melanoma. Cancer 100 (1): 122-9, 2004. [PUBMED Abstract]
Latest Updates to This Summary (05/16/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of intraocular melanoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Intraocular (Uveal) Melanoma Treatment is:
Shaheer A. Khan, DO (Columbia University Irving Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Intraocular (Uveal) Melanoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/eye/hp/intraocular-melanoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389482]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Leaving ChemoPrescribe
You are now leaving ChemoPrescribe website.
To return to the website, click on the back arrow on your browser.




 response by type of second cancer: sarcoma, skin cancer (BCC), meningioma, salivary gland cancer, glioma, breast cancer, and thyroid cancer.")

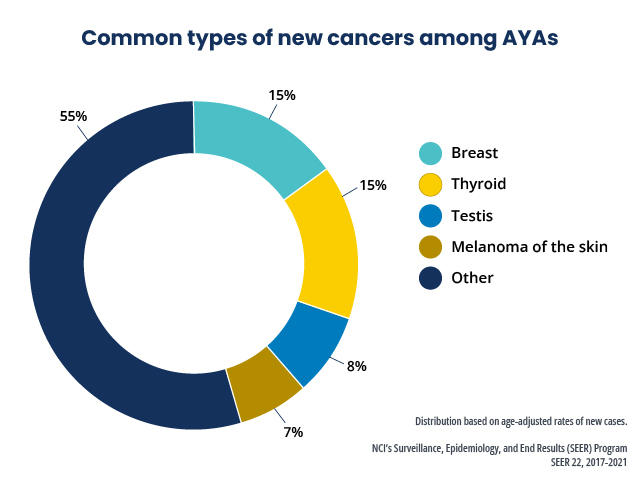







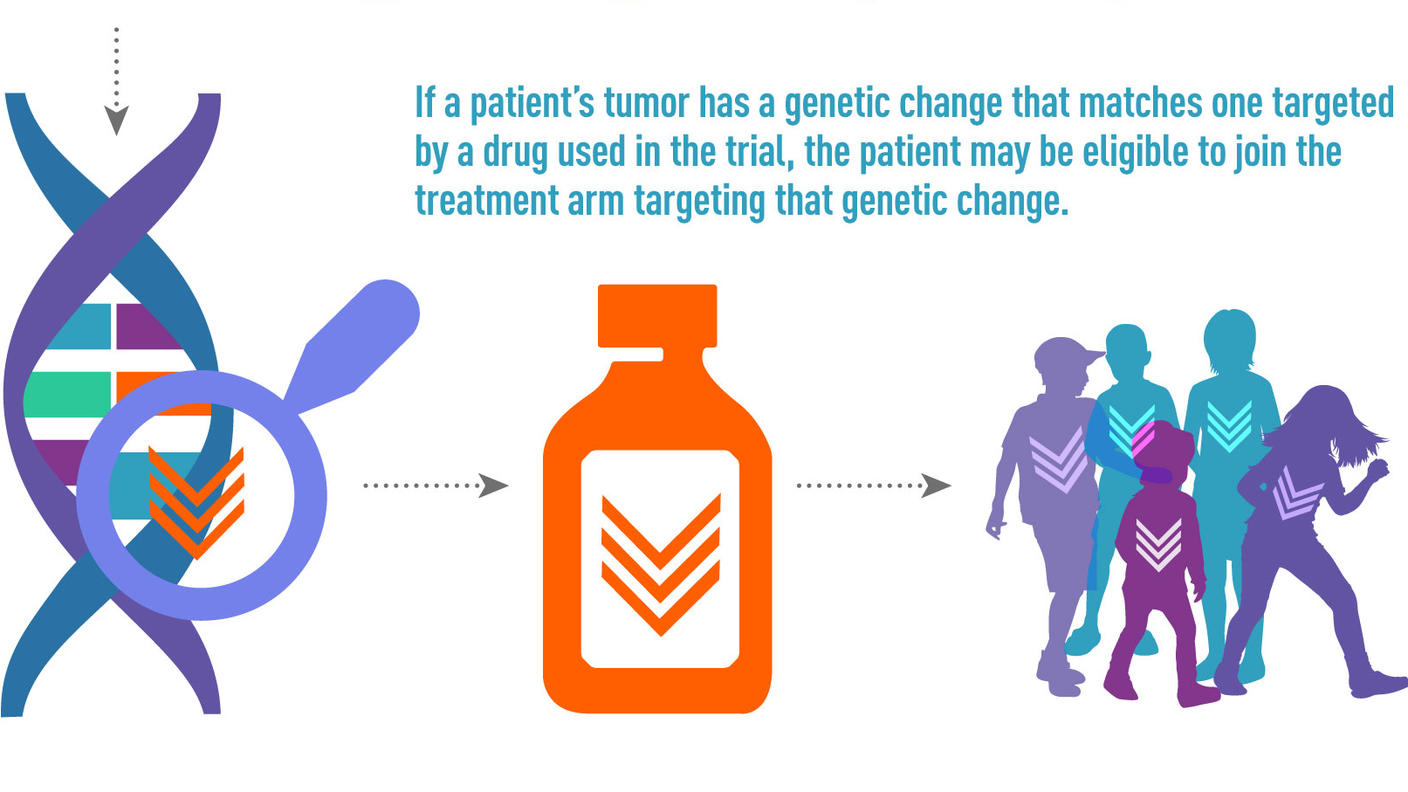

. An inset shows cancer cells spreading from the primary cancer, through the blood and lymph systems, to another part of the body where a metastatic tumor has formed.")
