AIDS-related lymphoma is a disease in which malignant (cancer) cells form in the lymph system of patients who have acquired immunodeficiency syndrome (AIDS).
There are many different types of lymphoma.
Signs of AIDS-related lymphoma include weight loss, fever, and drenching night sweats.
Tests that examine the lymph system and other parts of the body are used to diagnose AIDS-related lymphoma.
Certain factors affect prognosis (chance of recovery) and treatment options.
AIDS-related lymphoma is a disease in which malignant (cancer) cells form in the lymph system of patients who have acquired immunodeficiency syndrome (AIDS).
AIDS is caused by the human immunodeficiency virus (HIV), which attacks and weakens the body’s immune system. A weakened immune system is unable to fight infection and disease. People with HIV disease have an increased risk of infection and lymphoma or other types of cancer. A person with HIV and certain types of infection or cancer, such as lymphoma, is diagnosed as having AIDS. Sometimes, people are diagnosed with AIDS and AIDS-related lymphoma at the same time. For information about AIDS and its treatment, see the AIDSinfo website.
AIDS-related lymphoma is a type of cancer that affects the lymph system. The lymph system is part of the immune system. It helps protect the body from infection and disease.
Lymph vessels: A network of thin tubes that collect lymph from different parts of the body and return it to the bloodstream.
Lymph nodes: Small, bean-shaped structures that filter lymph and store white blood cells that help fight infection and disease. Lymph nodes are found along a network of lymph vessels throughout the body. Groups of lymph nodes are found in the neck, underarm, mediastinum, abdomen, pelvis, and groin.
Spleen: An organ that makes lymphocytes, stores red blood cells and lymphocytes, filters the blood, and destroys old blood cells. The spleen is on the left side of the abdomen near the stomach.
Thymus: An organ in which T lymphocytes mature and multiply. The thymus is in the chest behind the breastbone.
Tonsils: Two small masses of lymph tissue at the back of the throat. There is one tonsil on each side of the throat.
Bone marrow: The soft, spongy tissue in the center of certain bones, such as the hip bone and breastbone. White blood cells, red blood cells, and platelets are made in the bone marrow.
Lymph tissue is also found in other parts of the body such as the brain, stomach, thyroid gland, and skin.
EnlargeThe lymph system is part of the body’s immune system and is made up of tissues and organs that help protect the body from infection and disease. These include the tonsils, adenoids (not shown), thymus, spleen, bone marrow, lymph vessels, and lymph nodes. Lymph tissue is also found in many other parts of the body, including the small intestine.
Both non-Hodgkin lymphoma and Hodgkin lymphoma may occur in patients with AIDS, but non-Hodgkin lymphoma is more common. When a person with AIDS has non-Hodgkin lymphoma, it is called AIDS-related lymphoma. When AIDS-related lymphoma occurs in the central nervous system (CNS), it is called AIDS-related primary CNS lymphoma.
Non-Hodgkin lymphomas are grouped by the way their cells look under a microscope. They may be indolent (slow-growing) or aggressive (fast-growing). AIDS-related lymphomas are aggressive. There are two main types of AIDS-related non-Hodgkin lymphoma:
Signs of AIDS-related lymphoma include weight loss, fever, and drenching night sweats.
These and other signs and symptoms may be caused by AIDS-related lymphoma or by other conditions. Check with your doctor if you have any of the following:
Painless, swollen lymph nodes in the neck, chest, underarm, or groin.
A feeling of fullness below the ribs.
Tests that examine the lymph system and other parts of the body are used to diagnose AIDS-related lymphoma.
The following tests and procedures may be used:
Physical exam and health history: An exam of the body to check general signs of health, including checking for signs of disease, such as lumps or anything else that seems unusual. A history of the patient’s health, including fever, drenching night sweats, and weight loss, health habits, and past illnesses and treatments will also be taken.
Complete blood count (CBC): A procedure in which a sample of blood is drawn and checked for the following:
The number of red blood cells, white blood cells, and platelets.
The portion of the sample made up of red blood cells.
EnlargeComplete blood count (CBC). Blood is collected by inserting a needle into a vein and allowing the blood to flow into a tube. The blood sample is sent to the laboratory and the red blood cells, white blood cells, and platelets are counted. The CBC is used to test for, diagnose, and monitor many different conditions.
Blood chemistry studies: A procedure in which a blood sample is checked to measure the amounts of certain substances released into the blood by organs and tissues in the body. An unusual (higher or lower than normal) amount of a substance can be a sign of disease.
LDH test: A procedure in which a blood sample is checked to measure the amount of lactic dehydrogenase. An increased amount of LDH in the blood may be a sign of tissue damage, lymphoma, or other diseases.
Hepatitis B and hepatitis C test: A procedure in which a sample of blood is checked to measure the amounts of hepatitis B virus-specific antigens and/or antibodies and the amounts of hepatitis C virus-specific antibodies. These antigens or antibodies are called markers. Different markers or combinations of markers are used to determine whether a patient has a hepatitis B or C infection, has had a prior infection or vaccination, or is susceptible to infection.
HIV test: A test to measure the level of HIV antibodies in a sample of blood. Antibodies are made by the body when it is invaded by a foreign substance. A high level of HIV antibodies may mean the body has been infected with HIV.
CT scan (CAT scan): A procedure that makes a series of detailed pictures of areas inside the body, such as the neck, chest, abdomen, pelvis, and lymph nodes, taken from different angles. The pictures are made by a computer linked to an x-ray machine. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography.
PET scan (positron emission tomography scan): A procedure to find malignanttumor cells in the body. A small amount of radioactiveglucose (sugar) is injected into a vein. The PET scanner rotates around the body and makes a picture of where glucose is being used in the body. Malignant tumor cells show up brighter in the picture because they are more active and take up more glucose than normal cells do.
Bone marrow aspiration and biopsy: The removal of bone marrow and a small piece of bone by inserting a hollow needle into the hipbone or breastbone. A pathologist views the bone marrow and bone under a microscope to look for signs of cancer. EnlargeBone marrow aspiration and biopsy. After a small area of skin is numbed, a long, hollow needle is inserted through the patient’s skin and hip bone into the bone marrow. A sample of bone marrow and a small piece of bone are removed for examination under a microscope.
Lymph node biopsy: The removal of all or part of a lymph node. A pathologist views the tissue under a microscope to look for cancer cells. One of the following types of biopsies may be done:
Core biopsy: The removal of tissue from a lymph node using a wide needle.
Other areas of the body, such as the liver, lung, bone, bone marrow, and brain, may also have a sample of tissue removed and checked by a pathologist for signs of cancer.
If cancer is found, the following tests may be done to study the cancer cells:
Immunohistochemistry: A laboratory test that uses antibodies to check for certain antigens (markers) in a sample of a patient’s tissue. The antibodies are usually linked to an enzyme or a fluorescent dye. After the antibodies bind to a specific antigen in the tissue sample, the enzyme or dye is activated, and the antigen can then be seen under a microscope. This type of test is used to help diagnose cancer and to help tell one type of cancer from another type of cancer.
Cytogenetic analysis: A laboratory test in which the chromosomes of cells in a sample of blood or bone marrow are counted and checked for any changes, such as broken, missing, rearranged, or extra chromosomes. Changes in certain chromosomes may be a sign of cancer. Cytogenetic analysis is used to help diagnose cancer, plan treatment, or find out how well treatment is working.
FISH (fluorescence in situ hybridization): A laboratory test used to look at and count genes or chromosomes in cells and tissues. Pieces of DNA that contain fluorescent dyes are made in the laboratory and added to a sample of a patient’s cells or tissues. When these dyed pieces of DNA attach to certain genes or areas of chromosomes in the sample, they light up when viewed under a fluorescent microscope. The FISH test is used to help diagnose cancer and help plan treatment.
Immunophenotyping: A laboratory test that uses antibodies to identify cancer cells based on the types of antigens or markers on the surface of the cells. This test is used to help diagnose specific types of lymphoma.
Certain factors affect prognosis (chance of recovery) and treatment options.
The prognosis and treatment options depend on the following:
After AIDS-related lymphoma has been diagnosed, tests are done to find out if cancer cells have spread within the lymph system or to other parts of the body.
There are three ways that cancer spreads in the body.
The following stages are used for AIDS-related lymphoma:
Stage I
Stage II
Stage III
Stage IV
For treatment, AIDS-related lymphomas are grouped based on where they started in the body, as follows:
Peripheral/systemic lymphoma
Primary CNS lymphoma
After AIDS-related lymphoma has been diagnosed, tests are done to find out if cancer cells have spread within the lymph system or to other parts of the body.
The process used to find out if cancercells have spread within the lymph system or to other parts of the body is called staging. The information gathered from the staging process determines the stage of the disease. It is important to know the stage in order to plan treatment, but AIDS-relatedlymphoma is usually advanced when it is diagnosed.
The following tests and procedures may be used to find out if the cancer has spread:
MRI (magnetic resonance imaging) with gadolinium: A procedure that uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the body, such as the brain and spinal cord. A substance called gadolinium is injected into the patient through a vein. The gadolinium collects around the cancer cells so they show up brighter in the picture. This procedure is also called nuclear magnetic resonance imaging (NMRI).
Lumbar puncture: A procedure used to collect cerebrospinal fluid (CSF) from the spinal column. This is done by placing a needle between two bones in the spine and into the CSF around the spinal cord and removing a sample of the fluid. The sample of CSF is checked under a microscope for signs that the cancer has spread to the brain and spinal cord. The sample may also be checked for Epstein-Barr virus. This procedure is also called an LP or spinal tap. EnlargeLumbar puncture. A patient lies in a curled position on a table. After a small area on the lower back is numbed, a spinal needle (a long, thin needle) is inserted into the lower part of the spinal column to remove cerebrospinal fluid (CSF, shown in blue). The fluid may be sent to a laboratory for testing.
There are three ways that cancer spreads in the body.
Tissue. The cancer spreads from where it began by growing into nearby areas.
Lymph system. The cancer spreads from where it began by getting into the lymph system. The cancer travels through the lymph vessels to other parts of the body.
Blood. The cancer spreads from where it began by getting into the blood. The cancer travels through the blood vessels to other parts of the body.
The following stages are used for AIDS-related lymphoma:
Stage I
EnlargeStage I adult lymphoma. Cancer is found in one or more lymph nodes in a group of lymph nodes or, in rare cases, cancer is found in the Waldeyer’s ring, thymus, or spleen. In stage IE (not shown), cancer has spread to one area outside the lymph system.
In stage II, cancer is found in two or more groups of lymph nodes that are either above the diaphragm or below the diaphragm. EnlargeStage II adult lymphoma. Cancer is found in two or more groups of lymph nodes that are either above the diaphragm or below the diaphragm.
In stage IIE, cancer has spread from a group of lymph nodes to a nearby area that is outside the lymph system. Cancer may have spread to other lymph node groups on the same side of the diaphragm. EnlargeStage IIE adult lymphoma. Cancer has spread from a group of lymph nodes to a nearby area that is outside the lymph system. Cancer may have spread to other lymph node groups on the same side of the diaphragm.
In stage II, the term bulky disease refers to a larger tumor mass. The size of the tumor mass that is referred to as bulky disease varies based on the type of lymphoma.
Stage III
EnlargeStage III adult lymphoma. Cancer is found in groups of lymph nodes both above and below the diaphragm; or in a group of lymph nodes above the diaphragm and in the spleen.
in lymph nodes above the diaphragm and in the spleen.
Stage IV
EnlargeStage IV adult lymphoma. Cancer (a) has spread throughout one or more organs outside the lymph system; or (b) is found in two or more groups of lymph nodes that are either above the diaphragm or below the diaphragm and in one organ that is outside the lymph system and not near the affected lymph nodes; or (c) is found in groups of lymph nodes above the diaphragm and below the diaphragm and in any organ that is outside the lymph system; or (d) is found in the liver, bone marrow, more than one place in the lung, or cerebrospinal fluid (CSF). The cancer has not spread directly into the liver, bone marrow, lung, or CSF from nearby lymph nodes.
is found in two or more groups of lymph nodes that are either above the diaphragm or below the diaphragm and in one organ that is outside the lymph system and not near the affected lymph nodes; or
is found in groups of lymph nodes both above and below the diaphragm and in any organ that is outside the lymph system; or
is found in the liver, bone marrow, more than one place in the lung, or cerebrospinal fluid (CSF). The cancer has not spread directly into the liver, bone marrow, lung, or CSF from nearby lymph nodes.
Patients who are infected with the Epstein-Barr virus or whose AIDS-related lymphoma affects the bone marrow have an increased risk of the cancer spreading to the central nervous system (CNS).
For treatment, AIDS-related lymphomas are grouped based on where they started in the body, as follows:
Peripheral/systemic lymphoma
Lymphoma that starts in the lymph system or elsewhere in the body, other than the brain, is called peripheral/systemic lymphoma. It may spread throughout the body, including to the brain or bone marrow. It is often diagnosed in an advanced stage.
Primary CNS lymphoma
Primary CNS lymphoma starts in the central nervous system (brain and spinal cord). It is linked to the Epstein-Barr virus. Lymphoma that starts somewhere else in the body and spreads to the central nervous system is not primary CNS lymphoma.
Treatment Option Overview
Key Points
There are different types of treatment for patients with AIDS-related lymphoma.
Treatment of AIDS-related lymphoma combines treatment of the lymphoma with treatment for AIDS.
The following types of treatment are used:
Chemotherapy
Radiation therapy
High-dose chemotherapy with stem cell transplant
Targeted therapy
New types of treatment are being tested in clinical trials.
Treatment for AIDS-related lymphoma may cause side effects.
Patients may want to think about taking part in a clinical trial.
Patients can enter clinical trials before, during, or after starting their cancer treatment.
Follow-up tests may be needed.
There are different types of treatment for patients with AIDS-related lymphoma.
Different types of treatment are available for patients with AIDS-relatedlymphoma. Some treatments are standard (the currently used treatment), and some are being tested in clinical trials. A treatment clinical trial is a research study meant to help improve current treatments or obtain information on new treatments for patients with cancer. When clinical trials show that a new treatment is better than the standard treatment, the new treatment may become the standard treatment. Patients may want to think about taking part in a clinical trial. Some clinical trials are open only to patients who have not started treatment.
Treatment of AIDS-related lymphoma combines treatment of the lymphoma with treatment for AIDS.
Patients with AIDS have weakened immune systems and treatment can cause the immune system to become even weaker. For this reason, treating patients who have AIDS-related lymphoma is difficult and some patients may be treated with lower doses of drugs than lymphoma patients who do not have AIDS.
Highly active antiretroviral therapy (HAART) is used to lessen the damage to the immune system caused by HIV. Treatment with HAART may allow some patients with AIDS-related lymphoma to safely receive anticancer drugs in standard or higher doses. In these patients, treatment may work as well as it does in lymphoma patients who do not have AIDS. Medicine to prevent and treat infections, which can be serious, is also used.
For more information about AIDS and its treatment, see the AIDSinfo website.
Intrathecal chemotherapy may be used in patients who are more likely to have lymphoma in the central nervous system (CNS).
EnlargeIntrathecal chemotherapy. Anticancer drugs are injected into the intrathecal space, which is the space that holds the cerebrospinal fluid (CSF, shown in blue). There are two different ways to do this. One way, shown in the top part of the figure, is to inject the drugs into an Ommaya reservoir (a dome-shaped container that is placed under the scalp during surgery; it holds the drugs as they flow through a small tube into the brain). The other way, shown in the bottom part of the figure, is to inject the drugs directly into the CSF in the lower part of the spinal column, after a small area on the lower back is numbed.
Chemotherapy is used in the treatment of AIDS-related peripheral/systemic lymphoma. It is not yet known whether it is best to give HAART at the same time as chemotherapy or after chemotherapy ends.
Radiation therapy is a cancer treatment that uses high-energy x-rays or other types of radiation to kill cancer cells or keep them from growing. External radiation therapy uses a machine outside the body to send radiation toward the area of the body with cancer.
High-dose chemotherapy with stem cell transplant
High doses of chemotherapy are given to kill cancer cells. Healthy cells, including blood-forming cells, are also destroyed by the cancer treatment. Stem cell transplant is a treatment to replace the blood-forming cells. Stem cells (immature blood cells) are removed from the blood or bone marrow of the patient and are frozen and stored. After the patient completes chemotherapy, the stored stem cells are thawed and given back to the patient through an infusion. These reinfused stem cells grow into (and restore) the body’s blood cells.
Targeted therapy
Targeted therapy is a type of treatment that uses drugs or other substances to identify and attack specific cancer cells. Targeted therapies usually cause less harm to normal cells than chemotherapy or radiation therapy do.
Monoclonal antibodies: Monoclonal antibodies are immune system proteins made in the laboratory to treat many diseases, including cancer. As a cancer treatment, these antibodies can attach to a specific target on cancer cells or other cells that may help cancer cells grow. The antibodies are able to then kill the cancer cells, block their growth, or keep them from spreading. Monoclonal antibodies are given by infusion. These may be used alone or to carry drugs, toxins, or radioactive material directly to cancer cells. Rituximab is used in the treatment of AIDS-related peripheral/systemic lymphoma.
How do monoclonal antibodies work to treat cancer? This video shows how monoclonal antibodies, such as trastuzumab, pembrolizumab, and rituximab, block molecules cancer cells need to grow, flag cancer cells for destruction by the body’s immune system, or deliver harmful substances to cancer cells.
New types of treatment are being tested in clinical trials.
Information about clinical trials is available from the NCI website.
Treatment for AIDS-related lymphoma may cause side effects.
Patients may want to think about taking part in a clinical trial.
For some patients, taking part in a clinical trial may be the best treatment choice. Clinical trials are part of the cancer research process. Clinical trials are done to find out if new cancer treatments are safe and effective or better than the standard treatment.
Many of today’s standard treatments for cancer are based on earlier clinical trials. Patients who take part in a clinical trial may receive the standard treatment or be among the first to receive a new treatment.
Patients who take part in clinical trials also help improve the way cancer will be treated in the future. Even when clinical trials do not lead to effective new treatments, they often answer important questions and help move research forward.
Patients can enter clinical trials before, during, or after starting their cancer treatment.
Some clinical trials only include patients who have not yet received treatment. Other trials test treatments for patients whose cancer has not gotten better. There are also clinical trials that test new ways to stop cancer from recurring (coming back) or reduce the side effects of cancer treatment.
Clinical trials are taking place in many parts of the country. Information about clinical trials supported by NCI can be found on NCI’s clinical trials search webpage. Clinical trials supported by other organizations can be found on the ClinicalTrials.gov website.
Follow-up tests may be needed.
As you go through treatment, you will have follow-up tests or check-ups. Some tests that were done to diagnose or stage the cancer may be repeated to see how well the treatment is working. Decisions about whether to continue, change, or stop treatment may be based on the results of these tests.
Some of the tests will continue to be done from time to time after treatment has ended. The results of these tests can show if your condition has changed or if the cancer has recurred (come back).
Treatment of AIDS-Related Peripheral/Systemic Lymphoma
Use our clinical trial search to find NCI-supported cancer clinical trials that are accepting patients. You can search for trials based on the type of cancer, the age of the patient, and where the trials are being done. General information about clinical trials is also available.
Treatment of AIDS-Related Primary Central Nervous System Lymphoma
Use our clinical trial search to find NCI-supported cancer clinical trials that are accepting patients. You can search for trials based on the type of cancer, the age of the patient, and where the trials are being done. General information about clinical trials is also available.
To Learn More About AIDS-Related Lymphoma
For more information from the National Cancer Institute about AIDS-related lymphoma, see the following:
Physician Data Query (PDQ) is the National Cancer Institute’s (NCI’s) comprehensive cancer information database. The PDQ database contains summaries of the latest published information on cancer prevention, detection, genetics, treatment, supportive care, and complementary and alternative medicine. Most summaries come in two versions. The health professional versions have detailed information written in technical language. The patient versions are written in easy-to-understand, nontechnical language. Both versions have cancer information that is accurate and up to date and most versions are also available in Spanish.
PDQ is a service of the NCI. The NCI is part of the National Institutes of Health (NIH). NIH is the federal government’s center of biomedical research. The PDQ summaries are based on an independent review of the medical literature. They are not policy statements of the NCI or the NIH.
Purpose of This Summary
This PDQ cancer information summary has current information about the treatment of AIDS-related lymphoma. It is meant to inform and help patients, families, and caregivers. It does not give formal guidelines or recommendations for making decisions about health care.
Reviewers and Updates
Editorial Boards write the PDQ cancer information summaries and keep them up to date. These Boards are made up of experts in cancer treatment and other specialties related to cancer. The summaries are reviewed regularly and changes are made when there is new information. The date on each summary (“Updated”) is the date of the most recent change.
The information in this patient summary was taken from the health professional version, which is reviewed regularly and updated as needed, by the PDQ Adult Treatment Editorial Board.
Clinical Trial Information
A clinical trial is a study to answer a scientific question, such as whether one treatment is better than another. Trials are based on past studies and what has been learned in the laboratory. Each trial answers certain scientific questions in order to find new and better ways to help cancer patients. During treatment clinical trials, information is collected about the effects of a new treatment and how well it works. If a clinical trial shows that a new treatment is better than one currently being used, the new treatment may become “standard.” Patients may want to think about taking part in a clinical trial. Some clinical trials are open only to patients who have not started treatment.
Clinical trials can be found online at NCI’s website. For more information, call the Cancer Information Service (CIS), NCI’s contact center, at 1-800-4-CANCER (1-800-422-6237).
Permission to Use This Summary
PDQ is a registered trademark. The content of PDQ documents can be used freely as text. It cannot be identified as an NCI PDQ cancer information summary unless the whole summary is shown and it is updated regularly. However, a user would be allowed to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks in the following way: [include excerpt from the summary].”
The best way to cite this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ AIDS-Related Lymphoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/lymphoma/patient/aids-related-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389358]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use in the PDQ summaries only. If you want to use an image from a PDQ summary and you are not using the whole summary, you must get permission from the owner. It cannot be given by the National Cancer Institute. Information about using the images in this summary, along with many other images related to cancer can be found in Visuals Online. Visuals Online is a collection of more than 3,000 scientific images.
Disclaimer
The information in these summaries should not be used to make decisions about insurance reimbursement. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s E-mail Us.
Artist’s rendering of T cells (red and blue spheres) attacking cancer cells. T-cell therapy has been effective in treating certain lymphoma patients.
Credit: iStock
NCI-funded researchers are working to advance our understanding of how to treat lymphoma. All lymphomas start in the cells of the lymph system, which is part of the body’s immune system. Lymphomas are grouped into two main types: Hodgkin lymphoma and non-Hodgkin lymphoma (sometimes called NHL). But more than 70 different subtypes of the disease exist. Advances in understanding the gene changes that can lead to lymphoma are now helping scientists design more personalized treatments for these subtypes.
This page highlights some of the latest lymphoma research, including clinical advances that may soon translate into improved care and research findings from recent studies.
Treatment of Non-Hodgkin Lymphoma (NHL)
Most people diagnosed with lymphoma have a subtype of non-Hodgkin lymphoma. Non-Hodgkin lymphoma can either be aggressive or indolent.
Aggressive non-Hodgkin lymphoma grows and spreads quickly and usually requires immediate treatment. With modern treatment regimens, almost 70% of people with aggressive non-Hodgkin lymphoma will be considered cured. Research is now largely focused on finding better treatments for the minority of people with aggressive lymphoma who are not cured with initial therapy.
Indolent non-Hodgkin lymphoma grows slowly, and in some cases may not cause symptoms for years. People with indolent disease can often postpone treatment until their symptoms worsen, with no negative effects on survival. Sometimes, an indolent lymphoma can turn into aggressive lymphoma, which requires immediate treatment.
Indolent non-Hodgkin lymphoma largely cannot be cured with currently available therapies. The past two decades have seen improvements in extending the survival of people who are treated for this type of lymphoma. However, researchers are studying how to improve long-term survival further and working toward potentially curative treatments.
Chemotherapy, radiation therapy, targeted therapy, and immunotherapy are all used in the treatment of non-Hodgkin lymphoma. A stem cell transplant is sometimes used for lymphoma that has recurred, but this procedure has serious side effects. Four CAR T-cell therapies have been approved to treat some types of recurrent lymphoma. However, these newer therapies still can’t cure many people with recurrent lymphoma.
Most research on treatment for non-Hodgkin lymphoma is now focused on targeted therapy and immunotherapy. Researchers are also trying to identify gene changes in different types of lymphoma that might be targets for new drug development.
For example, in 2018, a study led by NCI researchers identified genetic subtypes of diffuse large B-cell lymphoma (the most common type of non-Hodgkin lymphoma) that could help explain why some patients with the disease respond to treatment and others don’t. Further studies may lead to more tailored treatments for patients with this type of lymphoma.
New targeted therapies
A signaling pathway is a series of chemical reactions that control one or more cell functions. Many types of non-Hodgkin lymphoma are driven by a signaling pathway called the B-cellreceptor signaling pathway. A drug called ibrutinib (Imbruvica) has been developed to shut down that pathway. It is being used and tested in a number of ways:
An early-phase NCI-supported study tested ibrutinib plus chemotherapy in people with primary central nervous system lymphoma, a very aggressive subtype of non-Hodgkin lymphoma. More than half of the patients in this small study went into complete, long-term remission.
However, in lymphoma, resistance to a single agent can develop quickly. Researchers are now testing combinations of targeted therapies to treat non-Hodgkin lymphoma to try to overcome resistance. For example, ongoing trials led by NCI researchers are testing a five-drug regimen and a six-drug regimen in people with aggressive or indolent B-cell lymphomas whose cancer has relapsed or is resistant to treatment.
Early results showed that the five-drug regimen, called ViPOR, shrank tumors substantially in about half of participants. Over a third of patients’ tumors disappeared entirely, called a complete response. Two years after treatment, most people who had a complete response remained in remission. These benefits were seen mainly in people with two specific subtypes of B-cell lymphoma.
Researchers are also trying to make standard treatment regimens less toxic. In one study, NCI researchers found that the intensity of standard chemotherapy could be reduced in adults with lower risk Burkitt lymphoma, an aggressive type of non-Hodgkin lymphoma, without compromising the potential for a cure.
Immunotherapy
Immunotherapy uses substances to stimulate or suppress the immune system to help the body fight cancer. Several immunotherapies have shown promise in treating different types of lymphoma.
CAR T cells. CAR T cells are a type of immunotherapy in which a patient’s T cells, a type of immune cell, are changed in the laboratory so they will better attack cancer cells. Four CAR T-cell therapies have been approved for the treatment of non-Hodgkin lymphoma:
Axicabtagene ciloleucel (Yescarta) for people with large B-cell lymphoma or follicular lymphoma whose cancer has progressed after receiving one prior treatment regimen.
Lisocabtagene maraleucel (Breyanzi) for people with some types of B-cell non-Hodgkin lymphoma that has relapsed or has not gotten better after at least two other treatments.
Early results from a phase 2 trial tested CAR T cells as initial therapy in people at very high risk of relapse showed that over three-quarters of patients had their cancer go into remission. However, long term results are not yet available and CAR T cells are not FDA approved in this setting.
CAR T cells are also being tested in other lymphoma subtypes, both aggressive and indolent, as well as in patients with lymphoma that has spread to the central nervous system.
Immunomodulating drugs. Immunomodulators are drugs that either stimulate or suppress the immune system. One such drug, lenalidomide (Revlimid), has been approved in combination with targeted therapies for previously treated follicular lymphoma and marginal zone lymphoma. It is also often used to treat diffuse large B-cell lymphoma.
Novel immunotherapies. Researchers are also testing novel ways to stimulate the immune system to fight lymphoma. For example, in 2018, a small trial showed that combining radiation therapy with the injection of a compound that stimulates the immune system could shrink some indolent B-cell lymphomas.
Immunotherapy drugs called bispecific antibodies are also under development. These drugs bind to lymphoma cells and the body’s own immune cells at the same time to bring them together. This allows the immune cells to kill the lymphoma cells. Five bispecific antibodies are in clinical trials for various types of lymphoma, including:
mosunetuzumab (Mosun), which triggered long-lasting remissions in almost 20% of people with aggressive B-cell non-Hodgkin lymphoma and almost 50% of people with indolent B-cell non-Hodgkin lymphoma in an early-phase clinical trial
Glofitamab, epcoritamab, and mosunetuzumab have all received accelerated approval from the FDA for the treatment of some lymphomas that have returned or gotten worse after at least two other treatments.
Hodgkin lymphoma is much less common than non-Hodgkin lymphoma. It is mostly seen in early adulthood (age 20–39) and in late adulthood (age 65 and older). More than 75% of all adults newly diagnosed with Hodgkin lymphoma can be cured with standard chemotherapy, radiation therapy, or both. Over the last 5 decades, deaths from Hodgkin lymphoma among adults have fallen more rapidly than deaths from any other cancer type.
Researchers are now focusing on adjusting standard treatment regimens to reduce the long-term side effects and improve quality of life for survivors. They are also testing better ways to treat the minority of patients whose cancer does recur.
Targeted therapies
A protein called CD30 is commonly found on the surface of Hodgkin lymphoma cells. A drug called brentuximab vedotin (Adcetris) that targets this protein has been approved as part of initial treatment for people with advanced Hodgkin lymphoma. Use of this new drug may help older patients avoid what had been the standard treatment with an especially toxic chemotherapy drug.
Clinical trials are now testing brentuximab vedotin combined with other chemotherapy drugs and with immunotherapies. The drug has also been approved by the FDA in combination with chemotherapy for some children and adolescents with Hodgkin lymphoma.
Immunotherapy
Immune checkpoint inhibitors that help T cells to better kill cancer cells have been effective in some people with recurrent Hodgkin lymphoma. Two such drugs—nivolumab (Opdivo) and pembrolizumab (Keytruda)—have been approved for some patients with Hodgkin lymphoma that has recurred after previous treatments.
Researchers are now testing these drugs in combination with other therapies, as well as earlier in treatment for some people with cancer that is likely to recur. For example, a large clinical trial recently found a benefit to giving nivolumab as part of initial treatment to teens and adults with advanced forms of classic Hodgkin lymphoma. More than 90% of trial participants who received nivolumab plus chemotherapy and targeted therapy were alive without their cancer starting to grow again 2 years after treatment, compared with 83% of those who received chemotherapy and targeted therapy alone
NCI-Supported Research Programs
The Lymphoma Specialized Programs of Research Excellence (Lymphoma SPOREs) are designed to quickly move basic scientific findings into clinical settings. The Lymphoma SPOREs support the development of new immunotherapies, novel targeted therapies, and new methods for determining prognosis for individual patients.
The goal of the International Lymphoma Epidemiology Consortium (InterLymph) is to enhance collaboration among epidemiologists studying lymphoma, provide a forum for the exchange of research ideas, and create a framework for collaborating on analyses that compile data from multiple studies.
The Lymphoma Epidemiology of Outcomes (LEO) Cohort Study was established to address the current and long-term health needs of non-Hodgkin lymphoma patients and survivors. The goal is to support a broad research agenda aimed at identifying novel clinical, epidemiologic, host, genetic, tumor, and treatment factors that significantly influence non-Hodgkin lymphoma prognosis and survivorship.
The Cancer Genome Characterization Initiative (CGCI) is supporting research to identify common gene changes in adult and pediatric cancers. Its results are freely available to the wider cancer research community, to spur the development of new targeted drugs. The HIV+ Tumor Molecular Characterization Project (HTMCP) and Burkitt Lymphoma Genome Sequencing Project (BLGSP) are two active CGCI projects.
The Clinical Trial Sequencing Project (CTSP) promotes the use of genomics in NCI-supported clinical trials. CTSP’s goal is to clarify the molecular basis of response and resistance to therapies studied. Diffuse large B-cell lymphoma is one of the cancer types under study, along with breast and renal cell carcinoma.
Clinical Trials
NCI funds and oversees both early- and late-phase clinical trials to develop new treatments and improve patient care. Trials are available for lymphoma treatment.
Lymphoma Research Results
The following are some of NCI’s latest news articles on lymphoma research:
Cutaneous T-cell lymphomas, which include mycosis fungoides and Sézary syndrome, are neoplasias of malignant T lymphocytes that usually possess the helper/inducer cell surface phenotype and initially present as skin involvement.[1] Cutaneous T-cell lymphomas should be distinguished from other T-cell lymphomas that involve the skin, such as anaplastic large cell lymphoma (CD30 positive), peripheral T-cell lymphoma (CD30 negative, with no epidermal involvement), or adult T-cell leukemia/lymphoma (usually with systemic involvement).[2,3] For more information about these types of T-cell lymphomas, see Peripheral T-Cell Non-Hodgkin Lymphoma Treatment.
Typically, the natural history of cutaneous T-cell lymphoma is indolent.[4] Symptoms of the disease may be present for long periods, in a range of 2 to 10 years, because cutaneous eruptions wax and wane before being confirmed by biopsy. Cutaneous T-cell lymphomas are treatable with available topical therapy, systemic therapy, or both. Curative modalities have proven elusive, with the possible exception of patients with minimal disease confined to the skin.
In addition, several benign or indolent conditions can be confused with mycosis fungoides. It is important to consult with a pathologist who has expertise in distinguishing these conditions.[1]
Prognosis and Survival
The prognosis of patients with cutaneous T-cell lymphomas is based on the extent of disease (stage) at presentation.[5] The presence of lymphadenopathy and involvement of peripheral blood and viscera increase in likelihood with worsening cutaneous involvement and define poor prognostic groups.[5–8] The Cutaneous Lymphoma International Consortium retrospectively reviewed 1,275 patients and found that the following four independent prognostic markers indicate a worse survival:[9]
Stage IV disease.
Age older than 60 years.
Large cell transformation.
Elevated lactate dehydrogenase.
The median survival following diagnosis varies according to stage. Patients with stage IA disease have a median survival of 20 years or more. Most deaths for this group are not caused by, nor are they related to, mycosis fungoides.[10,11] In contrast, more than 50% of patients with stage III through stage IV disease die of mycosis fungoides, with a median survival of approximately 5 years.[7,9,12,13] The Cutaneous Lymphoma International Prognostic index used male sex, age older than 60 years, plaques, lymph nodes, blood involvement, and visceral involvement as poor prognostic factors to define predicted overall survival (OS) and progression-free survival in both early-stage and advanced-stage groups.[14]
A report on 1,798 patients from the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) Program database found an increase in second malignancies in patients with mycosis fungoides (standardized incidence ratio, 1.32; 95% confidence interval [CI], 1.15–1.52), especially for Hodgkin lymphoma, non-Hodgkin lymphoma, and myeloma.[15] Another report on 4,459 patients from the SEER database found that the 19.2% of African American patients with mycosis fungoides had shorter OS, potentially attributable to disease characteristics, socioeconomic status, and type of therapy (hazard ratio, 1.47; 95% CI, 1.25–1.74; P < .001).[16]
Cutaneous disease can manifest as an eczematous patch or plaque stage covering less than 10% of the body surface (T1), a plaque stage covering 10% or more of the body surface (T2), or as tumors (T3) that frequently undergo necrotic ulceration.[17,18] Several retrospective studies showed that 20% of patients have disease that progresses from stage I or II to stage III or IV.[19–21] Sézary syndrome presents with generalized erythroderma (T4) and peripheral blood involvement. However, there is some disagreement about whether mycosis fungoides and Sézary syndrome are actually variants of the same disease.[22] The same retrospective study with a median follow-up of 14.5 years found that only 3% of 1,422 patients progressed from mycosis fungoides to Sézary syndrome.[19]
There is consensus that patients with Sézary syndrome (leukemic involvement) have a poor prognosis (median survival, 4 years), with or without the typical generalized erythroderma.[23,24] Cytologic transformation from a low-grade lymphoma to a high-grade lymphoma (large cell transformation) occurs rarely (<5%) during the course of these diseases and is associated with a poor prognosis.[25–27] A retrospective analysis of 100 cases with large cell transformation found reduced disease-specific survival with extracutaneous transformation, increased extent of skin lesions, and CD30 negativity.[28] A common cause of death during the tumor phase is septicemia caused by chronic skin infection with staph species, herpes simplex, herpes zoster, and fungal skin infections.[29,30]
Folliculotropic mycosis fungoides is a variant of mycosis fungoides marked by folliculotropic, rather than epidermotropic, neoplastic infiltrates, with preferential location in the head and neck area.[31] Early plaque-stage folliculotropic mycosis fungoides have a very indolent prognosis, while extracutaneous disease portends a very poor prognosis.[31]
References
Wilcox RA: Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol 92 (10): 1085-1102, 2017. [PUBMED Abstract]
Willemze R, Kerl H, Sterry W, et al.: EORTC classification for primary cutaneous lymphomas: a proposal from the Cutaneous Lymphoma Study Group of the European Organization for Research and Treatment of Cancer. Blood 90 (1): 354-71, 1997. [PUBMED Abstract]
Harris NL, Jaffe ES, Stein H, et al.: A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 84 (5): 1361-92, 1994. [PUBMED Abstract]
Diamandidou E, Cohen PR, Kurzrock R: Mycosis fungoides and Sezary syndrome. Blood 88 (7): 2385-409, 1996. [PUBMED Abstract]
Agar NS, Wedgeworth E, Crichton S, et al.: Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol 28 (31): 4730-9, 2010. [PUBMED Abstract]
Talpur R, Singh L, Daulat S, et al.: Long-term outcomes of 1,263 patients with mycosis fungoides and Sézary syndrome from 1982 to 2009. Clin Cancer Res 18 (18): 5051-60, 2012. [PUBMED Abstract]
Kim YH, Liu HL, Mraz-Gernhard S, et al.: Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol 139 (7): 857-66, 2003. [PUBMED Abstract]
Alberti-Violetti S, Talpur R, Schlichte M, et al.: Advanced-stage mycosis fungoides and Sézary syndrome: survival and response to treatment. Clin Lymphoma Myeloma Leuk 15 (6): e105-12, 2015. [PUBMED Abstract]
Scarisbrick JJ, Prince HM, Vermeer MH, et al.: Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J Clin Oncol 33 (32): 3766-73, 2015. [PUBMED Abstract]
Kim YH, Jensen RA, Watanabe GL, et al.: Clinical stage IA (limited patch and plaque) mycosis fungoides. A long-term outcome analysis. Arch Dermatol 132 (11): 1309-13, 1996. [PUBMED Abstract]
Vollmer RT: A review of survival in mycosis fungoides. Am J Clin Pathol 141 (5): 706-11, 2014. [PUBMED Abstract]
Zackheim HS, Amin S, Kashani-Sabet M, et al.: Prognosis in cutaneous T-cell lymphoma by skin stage: long-term survival in 489 patients. J Am Acad Dermatol 40 (3): 418-25, 1999. [PUBMED Abstract]
de Coninck EC, Kim YH, Varghese A, et al.: Clinical characteristics and outcome of patients with extracutaneous mycosis fungoides. J Clin Oncol 19 (3): 779-84, 2001. [PUBMED Abstract]
Benton EC, Crichton S, Talpur R, et al.: A cutaneous lymphoma international prognostic index (CLIPi) for mycosis fungoides and Sezary syndrome. Eur J Cancer 49 (13): 2859-68, 2013. [PUBMED Abstract]
Huang KP, Weinstock MA, Clarke CA, et al.: Second lymphomas and other malignant neoplasms in patients with mycosis fungoides and Sezary syndrome: evidence from population-based and clinical cohorts. Arch Dermatol 143 (1): 45-50, 2007. [PUBMED Abstract]
Su C, Nguyen KA, Bai HX, et al.: Racial disparity in mycosis fungoides: An analysis of 4495 cases from the US National Cancer Database. J Am Acad Dermatol 77 (3): 497-502.e2, 2017. [PUBMED Abstract]
Siegel RS, Pandolfino T, Guitart J, et al.: Primary cutaneous T-cell lymphoma: review and current concepts. J Clin Oncol 18 (15): 2908-25, 2000. [PUBMED Abstract]
Quaglino P, Pimpinelli N, Berti E, et al.: Time course, clinical pathways, and long-term hazards risk trends of disease progression in patients with classic mycosis fungoides: a multicenter, retrospective follow-up study from the Italian Group of Cutaneous Lymphomas. Cancer 118 (23): 5830-9, 2012. [PUBMED Abstract]
Wernham AG, Shah F, Amel-Kashipaz R, et al.: Stage I mycosis fungoides: frequent association with a favourable prognosis but disease progression and disease-specific mortality may occur. Br J Dermatol 173 (5): 1295-7, 2015. [PUBMED Abstract]
Desai M, Liu S, Parker S: Clinical characteristics, prognostic factors, and survival of 393 patients with mycosis fungoides and Sézary syndrome in the southeastern United States: a single-institution cohort. J Am Acad Dermatol 72 (2): 276-85, 2015. [PUBMED Abstract]
Olsen EA, Rook AH, Zic J, et al.: Sézary syndrome: immunopathogenesis, literature review of therapeutic options, and recommendations for therapy by the United States Cutaneous Lymphoma Consortium (USCLC). J Am Acad Dermatol 64 (2): 352-404, 2011. [PUBMED Abstract]
Kubica AW, Davis MD, Weaver AL, et al.: Sézary syndrome: a study of 176 patients at Mayo Clinic. J Am Acad Dermatol 67 (6): 1189-99, 2012. [PUBMED Abstract]
Thompson AK, Killian JM, Weaver AL, et al.: Sézary syndrome without erythroderma: A review of 16 cases at Mayo Clinic. J Am Acad Dermatol 76 (4): 683-688, 2017. [PUBMED Abstract]
Kim YH, Bishop K, Varghese A, et al.: Prognostic factors in erythrodermic mycosis fungoides and the Sézary syndrome. Arch Dermatol 131 (9): 1003-8, 1995. [PUBMED Abstract]
Arulogun SO, Prince HM, Ng J, et al.: Long-term outcomes of patients with advanced-stage cutaneous T-cell lymphoma and large cell transformation. Blood 112 (8): 3082-7, 2008. [PUBMED Abstract]
Kadin ME, Hughey LC, Wood GS: Large-cell transformation of mycosis fungoides-differential diagnosis with implications for clinical management: a consensus statement of the US Cutaneous Lymphoma Consortium. J Am Acad Dermatol 70 (2): 374-6, 2014. [PUBMED Abstract]
Benner MF, Jansen PM, Vermeer MH, et al.: Prognostic factors in transformed mycosis fungoides: a retrospective analysis of 100 cases. Blood 119 (7): 1643-9, 2012. [PUBMED Abstract]
Talpur R, Bassett R, Duvic M: Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and Sézary syndrome. Br J Dermatol 159 (1): 105-12, 2008. [PUBMED Abstract]
Lebas E, Arrese JE, Nikkels AF: Risk Factors for Skin Infections in Mycosis Fungoides. Dermatology 232 (6): 731-737, 2016. [PUBMED Abstract]
van Santen S, Roach RE, van Doorn R, et al.: Clinical Staging and Prognostic Factors in Folliculotropic Mycosis Fungoides. JAMA Dermatol 152 (9): 992-1000, 2016. [PUBMED Abstract]
Cellular Classification of Mycosis Fungoides and Other Cutaneous T-Cell Lymphomas
The histological diagnosis of mycosis fungoides and other cutaneous T-cell lymphomas is usually difficult to determine in the initial stages of the disease and may require the review of multiple biopsies by an experienced pathologist.
A definitive diagnosis from a skin biopsy requires the presence of cutaneous T-cell lymphoma cells (convoluted lymphocytes), a band-like upper dermal infiltrate, and epidermal infiltrations with Pautrier abscesses (collections of neoplastic lymphocytes). A definitive diagnosis of Sézary syndrome may be made from a peripheral blood evaluation when skin biopsies are consistent with the diagnosis. Supportive evidence for circulating Sézary cells is provided by T-cell receptor gene analysis, identification of the atypical lymphocytes with hyperconvoluted or cerebriform nuclei, and flow cytometry with the characteristic deletion of cell surface markers such as CD7 and CD26. However, none of these is individually pathognomonic for lymphoma.[1,2]
Established criteria for defining Sézary syndrome generally require identification of (1) a circulating T-cell clone by rearrangement of the T-cell receptor, plus (2) a level of Sézary cells in the blood of at least 1,000/μL.[3,4] It is less clear if other findings (such as a CD4:CD8 ratio ≥10, or the use of percentages instead of absolute numbers of atypical lymphocytes by flow cytometry) are reliable staging data in the absence of blood Sézary cell levels of at least 1,000/μL.[5]
References
Olsen EA, Rook AH, Zic J, et al.: Sézary syndrome: immunopathogenesis, literature review of therapeutic options, and recommendations for therapy by the United States Cutaneous Lymphoma Consortium (USCLC). J Am Acad Dermatol 64 (2): 352-404, 2011. [PUBMED Abstract]
Fraser-Andrews EA, Russell-Jones R, Woolford AJ, et al.: Diagnostic and prognostic importance of T-cell receptor gene analysis in patients with Sézary syndrome. Cancer 92 (7): 1745-52, 2001. [PUBMED Abstract]
Olsen E, Vonderheid E, Pimpinelli N, et al.: Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 110 (6): 1713-22, 2007. [PUBMED Abstract]
Olsen EA, Whittaker S, Willemze R, et al.: Primary cutaneous lymphoma: recommendations for clinical trial design and staging update from the ISCL, USCLC, and EORTC. Blood 140 (5): 419-437, 2022. [PUBMED Abstract]
Chrisman LP, Trimark PF, Pang Y, et al.: Updated cutaneous T-cell lymphoma TNMB staging criteria fail to identify patients with Sézary syndrome with low blood burden. Blood 144 (8): 914-917, 2024. [PUBMED Abstract]
Stage Information for Mycosis Fungoides and Other Cutaneous T-Cell Lymphomas
The American Joint Committee on Cancer (AJCC) has designated staging by TNM (tumor, node, metastasis) classification to define cutaneous T-cell lymphomas.[1] Peripheral blood involvement with cutaneous T-cell lymphoma cells is correlated with more advanced skin stage, lymph node and visceral involvement, and shortened survival.
Cutaneous T-cell lymphomas also have a formal staging system proposed by the International Society for Cutaneous Lymphomas and the European Organisation for Research and Treatment of Cancer.[2,3]
Table 1. Histopathological Staging of Lymph Nodes in Cutaneous T-Cell Lymphomaa
EORTC Classification
Dutch System
NCI-VA Classification
DL = dermatopathic lymphadenopathy; EORTC = European Organisation for Research and Treatment of Cancer; LN = lymph nodes; N = regional lymph node; NCI = National Cancer Institute; VA = U.S. Department of Veterans Affairs.
aReprinted with permission from AJCC: Primary Cutaneous Lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 967–72.
N1
Grade 1: DL
LN0: No atypical lymphocytes.
LN1: Occasional and isolated atypical lymphocytes (not arranged in clusters).
LN2: Many atypical lymphocytes or lymphocytes in 3-6‒cell clusters.
N2
Grade 2: DL; early involvement by mycosis fungoides (presence of cerebriform nuclei <7.5 µm [micrometer]).
LN3: Aggregates of atypical lymphocytes; nodal architecture preserved.
N3
Grade 3: Partial effacement of lymph node architecture; many atypical cerebriform mononuclear cells.
LN4: Partial/complete effacement of nodal architecture by atypical lymphocytes or frankly neoplastic cells.
Grade 4: Complete effacement.
Table 2. Definitions of TNM Stages IA and IBa
Stage
TNM
Description
B
Peripheral Blood Involvement Criteria
T = primary tumor; N = regional lymph node; M = distant metastasis; B = peripheral blood involvement.
aReprinted with permission from AJCC: Primary Cutaneous Lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 967–72.
The explanations for superscripts b through f are at the end of Table 5.
IA
T1, N0, M0
T1 = Limited patches,b papules, and/or plaquesc covering <10% of the skin surface.
B0,1
B0 = Absence of significant blood involvement: ≤5% of peripheral blood lymphocytes are atypical (Sézary) cells.d
–T1a = T1a (patch only).
–T1b = T1b (plaque ± patch).
–B0a = Clone negativee
–B0b = Clone positivee
N0 = No clinically abnormal peripheral lymph nodes;f biopsy not required.
B1 = Low blood tumor burden: >5% of peripheral blood lymphocytes are atypical (Sézary) cells, but does not meet the criteria of B2.
M0 = No visceral organ involvement.
–B1a = Clone negativee
–B1b = Clone positivee
IB
T2, N0, M0
T2 = Patches, papules, or plaques covering ≥10% of the skin surface.
B0,1
See B0, B1 descriptions above in this table, Stage IA.
–T2a = T2a (patch only).
–T2b = T2b (plaque ± patch).
N0 = No clinically abnormal peripheral lymph nodes;f biopsy not required.
M0 = No visceral organ involvement.
Table 3. Definitions of TNM Stages IIA and IIBa
Stage
TNM
Description
B
Peripheral Blood Involvement Criteria
T = primary tumor; N = regional lymph node; M = distant metastasis; B = peripheral blood involvement; LN = lymph nodes; NCI = National Cancer Institute.
aReprinted with permission from AJCC: Primary Cutaneous Lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 967–72.
The explanations for superscripts e through g are at the end of Table 5.
IIA
T1,2; N1,2; M0
See T1–2 descriptions above in Table 2, Stages IA, IB.
B0,1
See B0, B1 descriptions above in Table 2, Stage IA.
See B0, B1 descriptions above in Table 2, Stage IA.
–T3a = Multiple lesions involving 2 noncontiguous body regions.
–T3b = Multiple lesions involving ≥3 body regions.
N0 = No clinically abnormal peripheral lymph nodes;f biopsy not required.
See N1–2 descriptions above in this table, Stage IIA
M0 = No visceral organ involvement.
Table 4. Definitions of TNM Stages III, IIIA, and IIIBa
Stage
TNM
Description
B
Peripheral Blood Involvement Criteria
T = primary tumor; N = regional lymph node; M = distant metastasis; B = peripheral blood involvement.
aReprinted with permission from AJCC: Primary Cutaneous Lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 967–72.
III
T4, N0–2, M0
T4 = Confluence of erythema covering ≥80% of body surface area.
B0,1
See B0, B1 descriptions above in Table 2, Stage IA.
See N0–2 descriptions above in Table 3, Stages IIA, IIB.
M0 = No visceral organ involvement.
IIIA
T4, N0–2, M0
T4 = Confluence of erythema covering ≥80% of body surface area.
See N0–2 descriptions above in Table 3, Stages IIA, IIB.
M0 = No visceral organ involvement.
Table 5. Definitions of TNM Stages IVA1, IVA2, and IVBa
Stage
TNM
Description
B
Peripheral Blood Involvement Criteria
T = primary tumor; N = regional lymph node; M = distant metastasis; B = peripheral blood involvement; LN = lymph nodes; NCI = National Cancer Institute.
aReprinted with permission from AJCC: Primary Cutaneous Lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 967–72.
bFor skin, patch indicates any size skin lesion without significant elevation or induration. Presence/absence of hypo- or hyperpigmentation, scale, crusting, and/or poikiloderma should be noted.
cFor skin, plaque indicates any size skin lesion that is elevated or indurated. Presence/absence of scale, crusting, and/or poikiloderma should be noted. Histological features such as folliculotropism, large cell transformation (>25% large cells) and CD30 positivity or negativity, as well as clinical features such as ulceration, are important to document.
dFor blood, Sézary cells are defined as lymphocytes with hyperconvoluted cerebriform nuclei. If Sézary cells cannot be used to determine tumor burden for B2, then one of the following modified ISCL criteria, along with a positive clonal rearrangement of the T-cell receptor (TCR), may be used instead: (1) expanded CD4+ or CD3+ cells with a CD4/CD8 ratio of >10, or (2) expanded CD4+ cells with abnormal immunophenotype, including loss of CD7 (>40%) or CD26 (>30%).
eA T-cell clone is defined by polymerase chain reaction or Southern blot analysis of the TCR gene.
fFor node, abnormal peripheral lymph node(s) indicates any palpable peripheral node that on physical examination is firm, irregular, clustered, fixed or ≥1.5 cm in diameter. Node groups examined on physical examination include cervical, supraclavicular, epitrochlear, axillary, and inguinal. Central nodes, which generally are not amenable to pathological assessment, currently are not considered in the nodal classification unless used to establish N3 histopathologically.
gFor skin, tumor indicates at least one 1-cm diameter solid or nodular lesion with evidence of depth and/or vertical growth. Note the total number of lesions, total volume of lesions, largest size lesion, and region of body involved. Also note whether there is histological evidence of large cell transformation. Phenotyping for CD30 is encouraged.
hFor viscera, spleen and liver may be diagnosed by imaging criteria.
IVA1
T1–4, N0–2, M0
See T1‒2 descriptions above in Table 2, Stages IA, IB.
B2
B2 = High blood tumor burden: ≥1,000 mcg/L Sézary cellsd or >40% CD4+/CD7- or increased >30% CD4+/CD26- cells with positive clone.e
T3 = One or more tumorsg (≥1 cm in diameter).
–T3a = Multiple lesions involving 2 noncontiguous body regions.
–T3b = Multiple lesions involving ≥3 body regions.
T4 = Confluence of erythema covering ≥80% of body surface area.
See N0–2 descriptions above in Table 3, Stages IIA, IIB.
M0 = No visceral organ involvement.
IVA2
T1–4, N3, M0
See T1‒2 descriptions above in Table 2, Stages IA, IB and see T3–4 descriptions above in this table, Stage IVA1.
B0–2
See B0, B1 descriptions above in Table 2, Stage IA and see B2 description above in this table, Stage IVA1.
N3 = Clinically abnormal peripheral lymph nodes; histopathology Dutch grades 3–4 or NCI LN4; clone positive or negative.
M0 = No visceral organ involvement.
IVB
T1–4, N0–3, M1
See T1‒2 descriptions above in Table 2, Stages IA, IB and see T3–4 descriptions above in this table, Stage IVA1.
B0–2
See B0, B1 descriptions above in Table 2, Stage IA and see B2 description above in this table, Stage IVA1.
See N0–2 descriptions above in Table 3, Stages IIA, IIB.
N3 = Clinically abnormal peripheral lymph nodes; histopathology Dutch grades 3–4 or NCI LN4; clone positive or negative.
M1 = Visceral involvement (must have pathology confirmation,h and organ involved should be specified).
References
Primary cutaneous lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 967–72.
Olsen E, Vonderheid E, Pimpinelli N, et al.: Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 110 (6): 1713-22, 2007. [PUBMED Abstract]
Agar NS, Wedgeworth E, Crichton S, et al.: Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol 28 (31): 4730-9, 2010. [PUBMED Abstract]
Treatment Option Overview for Mycosis Fungoides and Other Cutaneous T-Cell Lymphomas
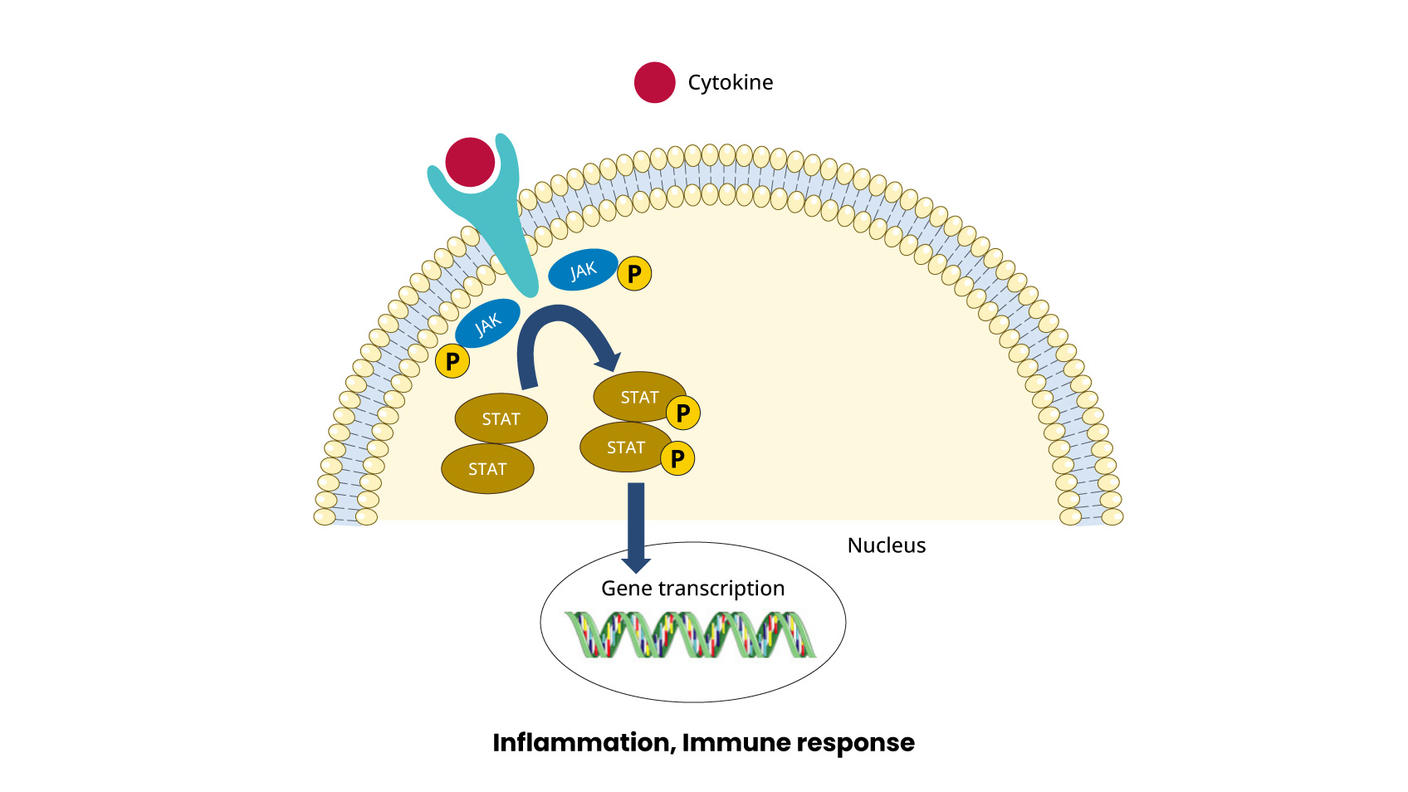
Anecdotal responses, some lasting for months, can be seen with aggressive antibiotic treatment of Staphylococcus aureus, with corresponding decreased expression of interleukin-2 receptors, STAT signaling, and T-cell proliferation.[1][Level of evidence C3]
These types of treatments produce remissions, but long-term remissions are uncommon. Treatment is considered palliative for most patients, although major symptomatic improvement is regularly achieved. Survival of more than 8 years is common for patients with early stages of disease. All patients with cutaneous T-cell lymphomas are candidates for clinical trials evaluating new approaches to treatment.
Table 6. Treatment Options for Mycosis Fungoides and Other Cutaneous T-Cell Lymphomas
Lindahl LM, Willerslev-Olsen A, Gjerdrum LMR, et al.: Antibiotics inhibit tumor and disease activity in cutaneous T-cell lymphoma. Blood 134 (13): 1072-1083, 2019. [PUBMED Abstract]
Treatment of Stage I and Stage II Mycosis Fungoides
Several forms of treatment can completely resolve skin lesions at stages I and II, so the choice of therapy depends on local expertise and the facilities available. With therapy, the survival of patients with stage IA disease can be expected to be the same as for age- and sex-matched controls.[1–3]
There is no curative therapy and no clear difference in overall survival (OS) among the treatment options for patients with stage I and stage II mycosis fungoides.
A randomized study of 103 patients compared combined total-skin electron-beam radiation (TSEB) plus combination chemotherapy with sequential topical therapies.[4] In the latter group, combination chemotherapy was reserved for patients with symptomatic extracutaneous disease or disease that was refractory to topical therapies. Patients with any disease stage were eligible. Although the complete response rate was higher with combined therapy, toxic effects were considerably greater, and no difference was seen in disease-free survival (DFS) or OS between the two groups.[4][Level of evidence A1]
Treatment Options for Stage I and Stage II Mycosis Fungoides
Treatment options for stages I and II mycosis fungoides include the following:[5]
Psoralen and ultraviolet A (PUVA) radiation therapy.[6–11]
Therapeutic trials with PUVA have shown an 80% to 90% complete remission rate in patients, with those in early cutaneous stages achieving the best responses. PUVA may be used in conjunction with systemic treatment.[10] Continued maintenance therapy with PUVA at more protracted intervals is generally required to prolong remission duration.[6–8,10] PUVA combined with interferon alfa-2a is associated with a high response rate.[9,10]
Single-arm and retrospective comparisons confirm the efficacy of narrowband ultraviolet B with 80% to 90% complete remission rates, especially for patients with early cutaneous stages.[12,13]
A Cochrane systematic review and meta-analysis compared PUVA with narrowband ultraviolet B radiation in 778 patients with early-stage mycosis fungoides (stage IA, IB, and IIA). Significantly higher complete responses were seen in patients treated with PUVA (73.8% vs. 62.2%; hazard ratio, 1.68; 95% confidence interval [CI], 1.02–2.76; P = .04). There were no significant differences in adverse effects.[11][Level of evidence B3]
Electron-beam radiation therapy of appropriate energies will penetrate only to the dermis, and thus, the skin alone can be treated without systemic effects. This therapy requires a radiation therapy facility with physics support and considerable technical expertise to deliver precise dosimetry. TSEB can result in short- and long-term cutaneous toxic effects and is not widely available.
This therapy can provide excellent palliation, with complete response rates as high as 80%, and may be combined with systemic treatment. Based on the long-term survival of these early-stage patients, electron-beam radiation therapy is sometimes used with curative intent.[14–18] Long-term DFS can be achieved in patients with unilesional mycosis fungoides treated with local radiation therapy.[19]
Local electron-beam radiation or orthovoltage radiation therapy may be used to palliate areas of bulky or symptomatic skin disease.[20,21]
Biological therapy
Interferon alfa or interferon gamma alone or in combination with topical therapy.[22,23][Level of evidence C3]
A retrospective review of 198 patients with mycosis fungoides and Sézary syndrome compared the time-to-next-treatment (TTNT) between patients who received interferon alfa and conventional chemotherapy. Interferon alfa provided a longer TTNT of 8.7 months (95% CI, 6.0–18.0) than did chemotherapy, with a TTNT of 3.9 months (95% CI, 3.2–5.1) (P < .00001).[23][Level of evidence C3]
Chemotherapy
Chemotherapeutic agents generally demonstrate short durations of response. In a retrospective review of 198 patients with advanced-stage disease, the median TTNT was 4 months.[23] However, these comparisons may be confounded by the order in which the agents were introduced.
Topical chemotherapy with mechlorethamine.[14,24,25]
This form of treatment may be used palliatively or to supplement therapeutic approaches directed against nodal or visceral disease. Topical application of mechlorethamine has produced regression of cutaneous lesions, with particular efficacy in early stages of disease. The overall complete remission rate is related to skin stage; 50% to 80% of TNM classification T1 patients, 25% to 75% of T2 patients, as many as 50% of T3 patients, and 20% to 40% of T4 patients have complete responses. The overall complete remission rate in 243 patients was 64% and was related to stage; as many as 35% of stage IV patients had complete responses. Treatments are usually continued for 2 to 3 years. Continuous 5-year DFS may be possible in as many as 33% of T1 patients.[14,24,25]
Fludarabine, cladribine, and pentostatin are active agents for mycosis fungoides.[30–33]
Single-agent chemotherapy or combination systemic chemotherapy (chlorambucil plus prednisone, mechlorethamine, cyclophosphamide, methotrexate) is often combined with treatment directed at the skin.[4,23,34,35]
Vorinostat or romidepsin or other histone deacetylase inhibitors (HDACi).[23,42–44][Level of evidence C3]
A retrospective review of 198 patients with mycosis fungoides and Sézary syndrome compared TTNT between HDACi and conventional chemotherapy. HDACi provided a longer TTNT of 4.5 months (95% CI, 4.0–6.1) than did chemotherapy, with a TTNT of 3.9 months (95% CI, 3.2–5.1; P = .01).[23][Level of evidence C3]
Two phase II trials of 58 patients with variable CD30 expression showed a 50% to 70% response rate with 50% of patients still in remission after 1 year.[45,46][Level of evidence C3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Kim YH, Jensen RA, Watanabe GL, et al.: Clinical stage IA (limited patch and plaque) mycosis fungoides. A long-term outcome analysis. Arch Dermatol 132 (11): 1309-13, 1996. [PUBMED Abstract]
Zackheim HS, Amin S, Kashani-Sabet M, et al.: Prognosis in cutaneous T-cell lymphoma by skin stage: long-term survival in 489 patients. J Am Acad Dermatol 40 (3): 418-25, 1999. [PUBMED Abstract]
Vollmer RT: A review of survival in mycosis fungoides. Am J Clin Pathol 141 (5): 706-11, 2014. [PUBMED Abstract]
Kaye FJ, Bunn PA, Steinberg SM, et al.: A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. N Engl J Med 321 (26): 1784-90, 1989. [PUBMED Abstract]
Trautinger F, Knobler R, Willemze R, et al.: EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome. Eur J Cancer 42 (8): 1014-30, 2006. [PUBMED Abstract]
Herrmann JJ, Roenigk HH, Hurria A, et al.: Treatment of mycosis fungoides with photochemotherapy (PUVA): long-term follow-up. J Am Acad Dermatol 33 (2 Pt 1): 234-42, 1995. [PUBMED Abstract]
Ramsay DL, Lish KM, Yalowitz CB, et al.: Ultraviolet-B phototherapy for early-stage cutaneous T-cell lymphoma. Arch Dermatol 128 (7): 931-3, 1992. [PUBMED Abstract]
Querfeld C, Rosen ST, Kuzel TM, et al.: Long-term follow-up of patients with early-stage cutaneous T-cell lymphoma who achieved complete remission with psoralen plus UV-A monotherapy. Arch Dermatol 141 (3): 305-11, 2005. [PUBMED Abstract]
Kuzel TM, Roenigk HH, Samuelson E, et al.: Effectiveness of interferon alfa-2a combined with phototherapy for mycosis fungoides and the Sézary syndrome. J Clin Oncol 13 (1): 257-63, 1995. [PUBMED Abstract]
Olsen EA, Hodak E, Anderson T, et al.: Guidelines for phototherapy of mycosis fungoides and Sézary syndrome: A consensus statement of the United States Cutaneous Lymphoma Consortium. J Am Acad Dermatol 74 (1): 27-58, 2016. [PUBMED Abstract]
Phan K, Ramachandran V, Fassihi H, et al.: Comparison of Narrowband UV-B With Psoralen-UV-A Phototherapy for Patients With Early-Stage Mycosis Fungoides: A Systematic Review and Meta-analysis. JAMA Dermatol 155 (3): 335-341, 2019. [PUBMED Abstract]
Almohideb M, Walsh S, Walsh S, et al.: Bath Psoralen-ultraviolet A and Narrowband Ultraviolet B Phototherapy as Initial Therapy for Early-stage Mycosis Fungoides: A Retrospective Cohort of 267 Cases at the University of Toronto. Clin Lymphoma Myeloma Leuk 17 (9): 604-612, 2017. [PUBMED Abstract]
Elcin G, Duman N, Karahan S, et al.: Long-term follow-up of early mycosis fungoides patients treated with narrowband ultraviolet B phototherapy. J Dermatolog Treat 25 (3): 268-73, 2014. [PUBMED Abstract]
Chinn DM, Chow S, Kim YH, et al.: Total skin electron beam therapy with or without adjuvant topical nitrogen mustard or nitrogen mustard alone as initial treatment of T2 and T3 mycosis fungoides. Int J Radiat Oncol Biol Phys 43 (5): 951-8, 1999. [PUBMED Abstract]
Quirós PA, Jones GW, Kacinski BM, et al.: Total skin electron beam therapy followed by adjuvant psoralen/ultraviolet-A light in the management of patients with T1 and T2 cutaneous T-cell lymphoma (mycosis fungoides). Int J Radiat Oncol Biol Phys 38 (5): 1027-35, 1997. [PUBMED Abstract]
Ysebaert L, Truc G, Dalac S, et al.: Ultimate results of radiation therapy for T1-T2 mycosis fungoides (including reirradiation). Int J Radiat Oncol Biol Phys 58 (4): 1128-34, 2004. [PUBMED Abstract]
Jones GW, Rosenthal D, Wilson LD: Total skin electron radiation for patients with erythrodermic cutaneous T-cell lymphoma (mycosis fungoides and the Sézary syndrome). Cancer 85 (9): 1985-95, 1999. [PUBMED Abstract]
Navi D, Riaz N, Levin YS, et al.: The Stanford University experience with conventional-dose, total skin electron-beam therapy in the treatment of generalized patch or plaque (T2) and tumor (T3) mycosis fungoides. Arch Dermatol 147 (5): 561-7, 2011. [PUBMED Abstract]
Micaily B, Miyamoto C, Kantor G, et al.: Radiotherapy for unilesional mycosis fungoides. Int J Radiat Oncol Biol Phys 42 (2): 361-4, 1998. [PUBMED Abstract]
Thomas TO, Agrawal P, Guitart J, et al.: Outcome of patients treated with a single-fraction dose of palliative radiation for cutaneous T-cell lymphoma. Int J Radiat Oncol Biol Phys 85 (3): 747-53, 2013. [PUBMED Abstract]
O’Malley JT, de Masson A, Lowry EL, et al.: Radiotherapy Eradicates Malignant T Cells and Is Associated with Improved Survival in Early-Stage Mycosis Fungoides. Clin Cancer Res 26 (2): 408-418, 2020. [PUBMED Abstract]
Olsen EA, Bunn PA: Interferon in the treatment of cutaneous T-cell lymphoma. Hematol Oncol Clin North Am 9 (5): 1089-107, 1995. [PUBMED Abstract]
Hughes CF, Khot A, McCormack C, et al.: Lack of durable disease control with chemotherapy for mycosis fungoides and Sézary syndrome: a comparative study of systemic therapy. Blood 125 (1): 71-81, 2015. [PUBMED Abstract]
Lessin SR, Duvic M, Guitart J, et al.: Topical chemotherapy in cutaneous T-cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatol 149 (1): 25-32, 2013. [PUBMED Abstract]
de Quatrebarbes J, Estève E, Bagot M, et al.: Treatment of early-stage mycosis fungoides with twice-weekly applications of mechlorethamine and topical corticosteroids: a prospective study. Arch Dermatol 141 (9): 1117-20, 2005. [PUBMED Abstract]
Zackheim HS, Kashani-Sabet M, McMillan A: Low-dose methotrexate to treat mycosis fungoides: a retrospective study in 69 patients. J Am Acad Dermatol 49 (5): 873-8, 2003. [PUBMED Abstract]
Wollina U, Dummer R, Brockmeyer NH, et al.: Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T-cell lymphoma. Cancer 98 (5): 993-1001, 2003. [PUBMED Abstract]
Dummer R, Quaglino P, Becker JC, et al.: Prospective international multicenter phase II trial of intravenous pegylated liposomal doxorubicin monochemotherapy in patients with stage IIB, IVA, or IVB advanced mycosis fungoides: final results from EORTC 21012. J Clin Oncol 30 (33): 4091-7, 2012. [PUBMED Abstract]
Quereux G, Marques S, Nguyen JM, et al.: Prospective multicenter study of pegylated liposomal doxorubicin treatment in patients with advanced or refractory mycosis fungoides or Sézary syndrome. Arch Dermatol 144 (6): 727-33, 2008. [PUBMED Abstract]
Saven A, Carrera CJ, Carson DA, et al.: 2-Chlorodeoxyadenosine: an active agent in the treatment of cutaneous T-cell lymphoma. Blood 80 (3): 587-92, 1992. [PUBMED Abstract]
Foss FM, Ihde DC, Breneman DL, et al.: Phase II study of pentostatin and intermittent high-dose recombinant interferon alfa-2a in advanced mycosis fungoides/Sézary syndrome. J Clin Oncol 10 (12): 1907-13, 1992. [PUBMED Abstract]
Foss FM, Ihde DC, Linnoila IR, et al.: Phase II trial of fludarabine phosphate and interferon alfa-2a in advanced mycosis fungoides/Sézary syndrome. J Clin Oncol 12 (10): 2051-9, 1994. [PUBMED Abstract]
Rosen ST, Foss FM: Chemotherapy for mycosis fungoides and the Sézary syndrome. Hematol Oncol Clin North Am 9 (5): 1109-16, 1995. [PUBMED Abstract]
Zackheim HS, Epstein EH: Low-dose methotrexate for the Sézary syndrome. J Am Acad Dermatol 21 (4 Pt 1): 757-62, 1989. [PUBMED Abstract]
Horwitz SM, Kim YH, Foss F, et al.: Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood 119 (18): 4115-22, 2012. [PUBMED Abstract]
Talpur R, Thompson A, Gangar P, et al.: Pralatrexate alone or in combination with bexarotene: long-term tolerability in relapsed/refractory mycosis fungoides. Clin Lymphoma Myeloma Leuk 14 (4): 297-304, 2014. [PUBMED Abstract]
Duffy R, Jennings T, Kartan S, et al.: Special Considerations in the Treatment of Mycosis Fungoides. Am J Clin Dermatol 20 (4): 571-578, 2019. [PUBMED Abstract]
Duvic M, Hymes K, Heald P, et al.: Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol 19 (9): 2456-71, 2001. [PUBMED Abstract]
Heald P, Mehlmauer M, Martin AG, et al.: Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma: results of the phase III clinical trial. J Am Acad Dermatol 49 (5): 801-15, 2003. [PUBMED Abstract]
Querfeld C, Rosen ST, Guitart J, et al.: Results of an open-label multicenter phase 2 trial of lenalidomide monotherapy in refractory mycosis fungoides and Sézary syndrome. Blood 123 (8): 1159-66, 2014. [PUBMED Abstract]
Duvic M, Dummer R, Becker JC, et al.: Panobinostat activity in both bexarotene-exposed and -naïve patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur J Cancer 49 (2): 386-94, 2013. [PUBMED Abstract]
Olsen EA, Kim YH, Kuzel TM, et al.: Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25 (21): 3109-15, 2007. [PUBMED Abstract]
Piekarz RL, Frye R, Turner M, et al.: Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 27 (32): 5410-7, 2009. [PUBMED Abstract]
Kim YH, Tavallaee M, Sundram U, et al.: Phase II Investigator-Initiated Study of Brentuximab Vedotin in Mycosis Fungoides and Sézary Syndrome With Variable CD30 Expression Level: A Multi-Institution Collaborative Project. J Clin Oncol 33 (32): 3750-8, 2015. [PUBMED Abstract]
Duvic M, Tetzlaff MT, Gangar P, et al.: Results of a Phase II Trial of Brentuximab Vedotin for CD30+ Cutaneous T-Cell Lymphoma and Lymphomatoid Papulosis. J Clin Oncol 33 (32): 3759-65, 2015. [PUBMED Abstract]
Treatment of Stage III and Stage IV Mycosis Fungoides and Sézary Syndrome
Mycosis Fungoides
There is no curative therapy and no clear difference in overall survival (OS) among the treatment options for patients with stage III and stage IV disease.
The use of single alkylating agents has produced objective responses in 60% of patients, with a duration of less than 6 months. One of the alkylating agents (e.g., mechlorethamine, cyclophosphamide, or chlorambucil) or the antimetabolite methotrexate is the most frequently used. Single agents have not cured any patients, and insufficient data exist to determine whether these agents prolong survival. Combination chemotherapy is not definitely superior to single agents. Even in patients with stage IV disease, treatments directed at the skin may provide significant palliation.
A randomized study of 103 patients compared combined total-skin electron-beam radiation (TSEB) plus combination chemotherapy with conservation therapy consisting of sequential topical therapies.[1] In the latter group, combination chemotherapy was reserved for patients with symptomatic extracutaneous disease or disease that was refractory to topical therapies. Patients with any disease stage were eligible. Although the complete response rate was higher with combined therapy, toxic effects were considerably greater, and no difference was seen in disease-free survival (DFS) or OS between the two groups.[1][Level of evidence A1]
Sézary Syndrome
Sézary syndrome is a rare leukemic variant of cutaneous T-cell lymphoma. It is characterized by erythroderma, circulating Sézary cells with cerebriform nuclei, lymphadenopathy, and pruritus.[2] This condition typically progresses rapidly, with only a short duration of response to most therapies. A retrospective review of 176 patients with Sézary syndrome identified the following poor prognostic factors:[3]
High lactate dehydrogenase.
Previous diagnosis of mycosis fungoides.
Presence of T-cell receptor gene rearrangements in skin, blood, or both.
Remissions attained by using extracorporeal photophoresis, interferon alfa, or retinoids may be followed by allogeneic stem cell transplant. In an anecdotal series of 16 patients with Sézary syndrome after allogeneic transplant, 9 were in complete remission after 4 years.[4]
Treatment Options for Stage III and Stage IV Mycosis Fungoides and Sézary Syndrome
Treatment options for stages III and IV mycosis fungoides and Sézary syndrome include the following (note that in this clinical setting, the skin is easily injured; any of the topical therapies must be administered with extreme caution):[2,5]
Psoralen and ultraviolet A (PUVA) radiation therapy.[6–11]
Therapeutic trials with PUVA have shown an 80% to 90% complete remission rate in patients, with those in early cutaneous stages achieving the best responses. PUVA may be used in conjunction with systemic treatment.[10] Continued maintenance therapy with PUVA at more protracted intervals is generally required to prolong remission duration.[6–8,10] PUVA combined with interferon alfa-2a is associated with a high response rate.[9,10]
Single-arm and retrospective comparisons confirm the efficacy of narrowband ultraviolet B with 80% to 90% complete remission rates, especially for patients with early cutaneous stages.[12,13]
A Cochrane systematic review and meta-analysis compared PUVA with narrowband ultraviolet B radiation in 778 patients with early-stage mycosis fungoides (stage IA, IB, and IIA). Significantly higher complete responses were seen in patients treated with PUVA (73.8% vs. 62.2%; hazard ratio, 1.68; 95% confidence interval [CI], 1.02–2.76; P = .04). There were no significant differences in adverse effects.[11][Level of evidence B3]
Extracorporeal photophoresis (ECP) alone [14–17] or in combination with TSEB.[18] ECP is particularly applicable for Sézary syndrome and erythrodermic mycosis fungoides.[17]
In a retrospective analysis of 65 patients, with a median follow-up of 48 months, use of ECP in the first to third line of treatment yielded a longer median time-to-next treatment (TTNT) than other systemic options (P < .03).[17][Level of evidence C3]
Electron-beam radiation therapy of appropriate energies will penetrate only to the dermis, and thus, the skin alone can be treated without systemic effects. This therapy requires a radiation therapy facility with physics support and considerable technical expertise to deliver precise dosimetry. TSEB can result in short- and long-term cutaneous toxic effects and is not widely available.
This therapy can provide excellent palliation, with complete response rates as high as 80%, and may be combined with systemic treatment. Based on the long-term survival of these early-stage patients, electron-beam radiation therapy is sometimes used with curative intent.[19–23] Long-term DFS can be achieved in patients with unilesional mycosis fungoides treated with local radiation therapy.[24]
Local electron-beam radiation or orthovoltage radiation therapy may be used to palliate areas of bulky or symptomatic disease.[25,26]
Biological therapy
Interferon alfa alone or in combination with other agents, such as topical therapy.[27,28]
A retrospective review of 198 patients with mycosis fungoides and Sézary syndrome compared the TTNT between patients who received interferon alfa and conventional chemotherapy. Interferon alfa provided a longer TTNT of 8.7 months (95% CI, 6.0–18.0) than did chemotherapy, with a TTNT of 3.9 months (95% CI, 3.2–5.1) (P < .00001).[29][Level of evidence C3]
Chemotherapy
Chemotherapeutic agents generally demonstrate short durations of response. In a retrospective review of 198 patients with advanced-stage disease, the median TTNT was 4 months.[29] However, these comparisons may be confounded by the order in which the agents were introduced.
Fludarabine, cladribine, and pentostatin are active agents for mycosis fungoides and Sézary syndrome.[27,29,31–33]
Single-agent chemotherapy or combination systemic chemotherapy (chlorambucil plus prednisone, mechlorethamine, cyclophosphamide, methotrexate) often combined with treatment directed at the skin.[1,29,34,35]
This form of treatment may be used palliatively or to supplement therapeutic approaches directed against nodal or visceral disease. Topical application of mechlorethamine has produced regression of cutaneous lesions, with particular efficacy in early stages of disease. The overall complete remission rate is related to skin stage; 50% to 80% of TNM classification T1 patients, 25% to 75% of T2 patients, as many as 50% of T3 patients, and 20% to 40% of T4 patients have complete responses. The overall complete remission rate in 243 patients was 64% and was related to stage; as many as 35% of stage IV patients had complete responses. Treatments are usually continued for 2 to 3 years. Continuous 5-year DFS may be possible in as many as 33% of T1 patients.[19,36,37]
Vorinostat or romidepsin or other histone deacetylase inhibitors (HDACi).[2,47–49]
A retrospective review of 198 patients with mycosis fungoides and Sézary syndrome compared TTNT between HDACi and conventional chemotherapy. HDACi provided a longer TTNT of 4.5 months (95% CI, 4.0–6.1) than did chemotherapy, with a TTNT of 3.9 months (95% CI, 3.2–5.1; P = .01).[29][Level of evidence C3]
Two phase II trials of 58 patients with variable CD30 expression showed a 50% to 70% response rate with 50% of patients still in remission after 1 year.[50,51][Level of evidence C3]
Anecdotal responses have been seen in patients with advanced relapsed or refractory mycosis fungoides. In a single-arm, multicenter, phase II trial of 24 patients treated with pembrolizumab, the overall response rate was 38%.[52][Level of evidence C3]
There are anecdotal reports of hyperprogression of T-cell malignancies following treatment with immune checkpoint inhibitors.[53,54]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Kaye FJ, Bunn PA, Steinberg SM, et al.: A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. N Engl J Med 321 (26): 1784-90, 1989. [PUBMED Abstract]
Olsen EA, Rook AH, Zic J, et al.: Sézary syndrome: immunopathogenesis, literature review of therapeutic options, and recommendations for therapy by the United States Cutaneous Lymphoma Consortium (USCLC). J Am Acad Dermatol 64 (2): 352-404, 2011. [PUBMED Abstract]
Kubica AW, Davis MD, Weaver AL, et al.: Sézary syndrome: a study of 176 patients at Mayo Clinic. J Am Acad Dermatol 67 (6): 1189-99, 2012. [PUBMED Abstract]
Polansky M, Talpur R, Daulat S, et al.: Long-Term Complete Responses to Combination Therapies and Allogeneic Stem Cell Transplants in Patients With Sézary Syndrome. Clin Lymphoma Myeloma Leuk 15 (5): e83-93, 2015. [PUBMED Abstract]
Trautinger F, Knobler R, Willemze R, et al.: EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome. Eur J Cancer 42 (8): 1014-30, 2006. [PUBMED Abstract]
Herrmann JJ, Roenigk HH, Hurria A, et al.: Treatment of mycosis fungoides with photochemotherapy (PUVA): long-term follow-up. J Am Acad Dermatol 33 (2 Pt 1): 234-42, 1995. [PUBMED Abstract]
Ramsay DL, Lish KM, Yalowitz CB, et al.: Ultraviolet-B phototherapy for early-stage cutaneous T-cell lymphoma. Arch Dermatol 128 (7): 931-3, 1992. [PUBMED Abstract]
Querfeld C, Rosen ST, Kuzel TM, et al.: Long-term follow-up of patients with early-stage cutaneous T-cell lymphoma who achieved complete remission with psoralen plus UV-A monotherapy. Arch Dermatol 141 (3): 305-11, 2005. [PUBMED Abstract]
Kuzel TM, Roenigk HH, Samuelson E, et al.: Effectiveness of interferon alfa-2a combined with phototherapy for mycosis fungoides and the Sézary syndrome. J Clin Oncol 13 (1): 257-63, 1995. [PUBMED Abstract]
Olsen EA, Hodak E, Anderson T, et al.: Guidelines for phototherapy of mycosis fungoides and Sézary syndrome: A consensus statement of the United States Cutaneous Lymphoma Consortium. J Am Acad Dermatol 74 (1): 27-58, 2016. [PUBMED Abstract]
Phan K, Ramachandran V, Fassihi H, et al.: Comparison of Narrowband UV-B With Psoralen-UV-A Phototherapy for Patients With Early-Stage Mycosis Fungoides: A Systematic Review and Meta-analysis. JAMA Dermatol 155 (3): 335-341, 2019. [PUBMED Abstract]
Almohideb M, Walsh S, Walsh S, et al.: Bath Psoralen-ultraviolet A and Narrowband Ultraviolet B Phototherapy as Initial Therapy for Early-stage Mycosis Fungoides: A Retrospective Cohort of 267 Cases at the University of Toronto. Clin Lymphoma Myeloma Leuk 17 (9): 604-612, 2017. [PUBMED Abstract]
Elcin G, Duman N, Karahan S, et al.: Long-term follow-up of early mycosis fungoides patients treated with narrowband ultraviolet B phototherapy. J Dermatolog Treat 25 (3): 268-73, 2014. [PUBMED Abstract]
Edelson R, Berger C, Gasparro F, et al.: Treatment of cutaneous T-cell lymphoma by extracorporeal photochemotherapy. Preliminary results. N Engl J Med 316 (6): 297-303, 1987. [PUBMED Abstract]
Heald PW, Perez MI, McKiernan G, et al.: Extracorporeal photochemotherapy for CTCL. Prog Clin Biol Res 337: 443-7, 1990. [PUBMED Abstract]
Scarisbrick JJ, Taylor P, Holtick U, et al.: U.K. consensus statement on the use of extracorporeal photopheresis for treatment of cutaneous T-cell lymphoma and chronic graft-versus-host disease. Br J Dermatol 158 (4): 659-78, 2008. [PUBMED Abstract]
Gao C, McCormack C, van der Weyden C, et al.: Prolonged survival with the early use of a novel extracorporeal photopheresis regimen in patients with Sézary syndrome. Blood 134 (16): 1346-1350, 2019. [PUBMED Abstract]
Palareti G, Maccaferri M, Manotti C, et al.: Fibrinogen assays: a collaborative study of six different methods. C.I.S.M.E.L. Comitato Italiano per la Standardizzazione dei Metodi in Ematologia e Laboratorio. Clin Chem 37 (5): 714-9, 1991. [PUBMED Abstract]
Chinn DM, Chow S, Kim YH, et al.: Total skin electron beam therapy with or without adjuvant topical nitrogen mustard or nitrogen mustard alone as initial treatment of T2 and T3 mycosis fungoides. Int J Radiat Oncol Biol Phys 43 (5): 951-8, 1999. [PUBMED Abstract]
Quirós PA, Jones GW, Kacinski BM, et al.: Total skin electron beam therapy followed by adjuvant psoralen/ultraviolet-A light in the management of patients with T1 and T2 cutaneous T-cell lymphoma (mycosis fungoides). Int J Radiat Oncol Biol Phys 38 (5): 1027-35, 1997. [PUBMED Abstract]
Ysebaert L, Truc G, Dalac S, et al.: Ultimate results of radiation therapy for T1-T2 mycosis fungoides (including reirradiation). Int J Radiat Oncol Biol Phys 58 (4): 1128-34, 2004. [PUBMED Abstract]
Jones GW, Rosenthal D, Wilson LD: Total skin electron radiation for patients with erythrodermic cutaneous T-cell lymphoma (mycosis fungoides and the Sézary syndrome). Cancer 85 (9): 1985-95, 1999. [PUBMED Abstract]
Navi D, Riaz N, Levin YS, et al.: The Stanford University experience with conventional-dose, total skin electron-beam therapy in the treatment of generalized patch or plaque (T2) and tumor (T3) mycosis fungoides. Arch Dermatol 147 (5): 561-7, 2011. [PUBMED Abstract]
Micaily B, Miyamoto C, Kantor G, et al.: Radiotherapy for unilesional mycosis fungoides. Int J Radiat Oncol Biol Phys 42 (2): 361-4, 1998. [PUBMED Abstract]
Thomas TO, Agrawal P, Guitart J, et al.: Outcome of patients treated with a single-fraction dose of palliative radiation for cutaneous T-cell lymphoma. Int J Radiat Oncol Biol Phys 85 (3): 747-53, 2013. [PUBMED Abstract]
O’Malley JT, de Masson A, Lowry EL, et al.: Radiotherapy Eradicates Malignant T Cells and Is Associated with Improved Survival in Early-Stage Mycosis Fungoides. Clin Cancer Res 26 (2): 408-418, 2020. [PUBMED Abstract]
Foss FM, Ihde DC, Breneman DL, et al.: Phase II study of pentostatin and intermittent high-dose recombinant interferon alfa-2a in advanced mycosis fungoides/Sézary syndrome. J Clin Oncol 10 (12): 1907-13, 1992. [PUBMED Abstract]
Olsen EA, Bunn PA: Interferon in the treatment of cutaneous T-cell lymphoma. Hematol Oncol Clin North Am 9 (5): 1089-107, 1995. [PUBMED Abstract]
Hughes CF, Khot A, McCormack C, et al.: Lack of durable disease control with chemotherapy for mycosis fungoides and Sézary syndrome: a comparative study of systemic therapy. Blood 125 (1): 71-81, 2015. [PUBMED Abstract]
Zackheim HS, Kashani-Sabet M, McMillan A: Low-dose methotrexate to treat mycosis fungoides: a retrospective study in 69 patients. J Am Acad Dermatol 49 (5): 873-8, 2003. [PUBMED Abstract]
Saven A, Carrera CJ, Carson DA, et al.: 2-Chlorodeoxyadenosine: an active agent in the treatment of cutaneous T-cell lymphoma. Blood 80 (3): 587-92, 1992. [PUBMED Abstract]
Foss FM, Ihde DC, Linnoila IR, et al.: Phase II trial of fludarabine phosphate and interferon alfa-2a in advanced mycosis fungoides/Sézary syndrome. J Clin Oncol 12 (10): 2051-9, 1994. [PUBMED Abstract]
Rosen ST, Foss FM: Chemotherapy for mycosis fungoides and the Sézary syndrome. Hematol Oncol Clin North Am 9 (5): 1109-16, 1995. [PUBMED Abstract]
Zackheim HS, Epstein EH: Low-dose methotrexate for the Sézary syndrome. J Am Acad Dermatol 21 (4 Pt 1): 757-62, 1989. [PUBMED Abstract]
Lessin SR, Duvic M, Guitart J, et al.: Topical chemotherapy in cutaneous T-cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatol 149 (1): 25-32, 2013. [PUBMED Abstract]
de Quatrebarbes J, Estève E, Bagot M, et al.: Treatment of early-stage mycosis fungoides with twice-weekly applications of mechlorethamine and topical corticosteroids: a prospective study. Arch Dermatol 141 (9): 1117-20, 2005. [PUBMED Abstract]
Dummer R, Quaglino P, Becker JC, et al.: Prospective international multicenter phase II trial of intravenous pegylated liposomal doxorubicin monochemotherapy in patients with stage IIB, IVA, or IVB advanced mycosis fungoides: final results from EORTC 21012. J Clin Oncol 30 (33): 4091-7, 2012. [PUBMED Abstract]
Wollina U, Dummer R, Brockmeyer NH, et al.: Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T-cell lymphoma. Cancer 98 (5): 993-1001, 2003. [PUBMED Abstract]
Quereux G, Marques S, Nguyen JM, et al.: Prospective multicenter study of pegylated liposomal doxorubicin treatment in patients with advanced or refractory mycosis fungoides or Sézary syndrome. Arch Dermatol 144 (6): 727-33, 2008. [PUBMED Abstract]
Horwitz SM, Kim YH, Foss F, et al.: Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood 119 (18): 4115-22, 2012. [PUBMED Abstract]
Talpur R, Thompson A, Gangar P, et al.: Pralatrexate alone or in combination with bexarotene: long-term tolerability in relapsed/refractory mycosis fungoides. Clin Lymphoma Myeloma Leuk 14 (4): 297-304, 2014. [PUBMED Abstract]
Duffy R, Jennings T, Kartan S, et al.: Special Considerations in the Treatment of Mycosis Fungoides. Am J Clin Dermatol 20 (4): 571-578, 2019. [PUBMED Abstract]
Querfeld C, Rosen ST, Guitart J, et al.: Results of an open-label multicenter phase 2 trial of lenalidomide monotherapy in refractory mycosis fungoides and Sézary syndrome. Blood 123 (8): 1159-66, 2014. [PUBMED Abstract]
Duvic M, Hymes K, Heald P, et al.: Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol 19 (9): 2456-71, 2001. [PUBMED Abstract]
Heald P, Mehlmauer M, Martin AG, et al.: Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma: results of the phase III clinical trial. J Am Acad Dermatol 49 (5): 801-15, 2003. [PUBMED Abstract]
Duvic M, Dummer R, Becker JC, et al.: Panobinostat activity in both bexarotene-exposed and -naïve patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur J Cancer 49 (2): 386-94, 2013. [PUBMED Abstract]
Olsen EA, Kim YH, Kuzel TM, et al.: Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25 (21): 3109-15, 2007. [PUBMED Abstract]
Piekarz RL, Frye R, Turner M, et al.: Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 27 (32): 5410-7, 2009. [PUBMED Abstract]
Kim YH, Tavallaee M, Sundram U, et al.: Phase II Investigator-Initiated Study of Brentuximab Vedotin in Mycosis Fungoides and Sézary Syndrome With Variable CD30 Expression Level: A Multi-Institution Collaborative Project. J Clin Oncol 33 (32): 3750-8, 2015. [PUBMED Abstract]
Duvic M, Tetzlaff MT, Gangar P, et al.: Results of a Phase II Trial of Brentuximab Vedotin for CD30+ Cutaneous T-Cell Lymphoma and Lymphomatoid Papulosis. J Clin Oncol 33 (32): 3759-65, 2015. [PUBMED Abstract]
Khodadoust MS, Rook AH, Porcu P, et al.: Pembrolizumab in Relapsed and Refractory Mycosis Fungoides and Sézary Syndrome: A Multicenter Phase II Study. J Clin Oncol 38 (1): 20-28, 2020. [PUBMED Abstract]
Ratner L, Waldmann TA, Janakiram M, et al.: Rapid Progression of Adult T-Cell Leukemia-Lymphoma after PD-1 Inhibitor Therapy. N Engl J Med 378 (20): 1947-1948, 2018. [PUBMED Abstract]
Bennani NN, Kim HJ, Pederson LD, et al.: Nivolumab in patients with relapsed or refractory peripheral T-cell lymphoma: modest activity and cases of hyperprogression. J Immunother Cancer 10 (6): , 2022. [PUBMED Abstract]
Treatment of Recurrent Mycosis Fungoides and Sézary Syndrome
The treatment of patients with relapsed mycosis fungoides and Sézary syndrome involves the joint decisions of a dermatologist, medical oncologist, and radiation oncologist. It may be possible to re-treat localized areas of relapse in the skin with additional electron-beam radiation therapy or to repeat total-skin electron-beam radiation therapy (TSEB).[1] Photon radiation to bulky skin or nodal masses may prove beneficial. If these options are not possible, then continued topical treatment with other modalities, such as mechlorethamine or psoralen and ultraviolet A radiation (PUVA) may be warranted to relieve cutaneous symptoms.
Patients should consider clinical trials as a therapeutic option.
Treatment Options for Recurrent Mycosis Fungoides and Sézary Syndrome
Treatment options under clinical evaluation for recurrent mycosis fungoides and Sézary syndrome include the following:[2,3]
Additional electron-beam radiation therapy or a repeat of TSEB.
Electron-beam radiation therapy of appropriate energies will penetrate only to the dermis, and thus, the skin alone can be treated without systemic effects. This therapy requires a radiation therapy facility with physics support and considerable technical expertise to deliver precise dosimetry. TSEB can result in short- and long-term cutaneous toxic effects and is not widely available.
This therapy can provide excellent palliation, with complete response rates as high as 80%, and may be combined with systemic treatment. Based on the long-term survival of these early-stage patients, electron-beam radiation therapy is sometimes used with curative intent.[4–8] Long-term disease-free survival (DFS) can be achieved in patients with unilesional mycosis fungoides treated with local radiation therapy.[9]
Photon radiation to bulky skin or nodal masses.[10]
Therapeutic trials with PUVA have shown an 80% to 90% complete remission rate in patients, with those in early cutaneous stages achieving the best responses. PUVA may be used in conjunction with systemic treatment.[15] Continued maintenance therapy with PUVA at more protracted intervals is generally required to prolong remission duration.[11–13,15] PUVA combined with interferon alfa-2a is associated with a high response rate.[14,15]
Single-arm and retrospective comparisons confirm the efficacy of narrowband ultraviolet B with 80% to 90% complete remission rates, especially for patients with early cutaneous stages.[17,18]
A Cochrane systematic review and meta-analysis compared PUVA with narrowband ultraviolet B radiation in 778 patients with early-stage mycosis fungoides (stage IA, IB, and IIA). Significantly higher complete response rates were seen in patients treated with PUVA (73.8% vs. 62.2%; hazard ratio [HR], 1.68; 95% confidence interval [CI], 1.02–2.76; P = .04). There were no significant differences in adverse effects.[16][Level of evidence B3]
Extracorporeal photophoresis (ECP) has produced tumor regression in patients who are resistant to other therapies.[19,20]
In a retrospective analysis of 65 patients, with a median follow-up of 48 months, use of ECP in the first to third line of treatment yielded a longer median time-to-next treatment (TTNT) than other systemic options (P < .03).[21][Level of evidence C3]
Chemotherapy
Topical treatment with mechlorethamine or PUVA.
This form of treatment may be used palliatively or to supplement therapeutic approaches directed against nodal or visceral disease. Topical application of mechlorethamine has produced regression of cutaneous lesions, with particular efficacy in early stages of disease. The overall complete remission rate is related to skin stage; 50% to 80% of TNM classification T1 patients, 25% to 75% of T2 patients, as many as 50% of T3 patients, and 20% to 40% of T4 patients have complete responses. The overall complete remission rate in 243 patients was 64% and was related to stage; as many as 35% of stage IV patients had complete responses. Treatments are usually continued for 2 to 3 years. Continuous 5-year DFS may be possible in as many as 33% of T1 patients.[4,22,23]
Chemotherapeutic agents generally yield short durations of response. In a retrospective review of 198 patients with advanced-stage disease, the median TTNT was 4 months.[26]
Vorinostat or romidepsin or other histone deacetylase inhibitors (HDACi).[36–38]
A retrospective review of 198 patients with mycosis fungoides and Sézary syndrome compared the TTNT between HDACi and conventional chemotherapy. HDACi provided a longer TTNT of 4.5 months (95% CI, 4.0–6.1) than did chemotherapy, with a TTNT of 3.9 months (95% CI, 3.2–5.1; P = .01).[26][Level of evidence C3]
Biological therapy
Interferon alfa alone or in combination with other agents, such as topical therapy.[39,40]
A retrospective review of 198 patients with mycosis fungoides and Sézary syndrome compared the TTNT between patients who received interferon alfa and conventional chemotherapy. Interferon alfa provided a longer TTNT of 8.7 months (95% CI, 6.0–18.0) than did chemotherapy, with a TTNT of 3.9 months (95% CI, 3.2–5.1) (P < .00001).[26][Level of evidence C3]
Two phase II trials of 58 patients with variable CD30 expression showed a 50% to 70% response rate with 50% of patients still in remission after 1 year.[46,47][Level of evidence C3]
In a prospective randomized trial, 372 previously treated patients received either mogamulizumab, a monoclonal antibody directed against C-C chemokine receptor 4, or the HDACi vorinostat.
With a median follow-up of 17 months, the median progression-free survival was 7.7 months for patients who received mogamulizumab and 3.1 months for patients who received vorinostat (HR, 0.53; 95% CI, 0.41−0.69; P < .0001).[48][Level of evidence B1]
Mogamulizumab appeared to be especially effective in patients with blood involvement, such as those with Sézary syndrome.
This preliminary study was not designed to detect differences in OS.
Mogamulizumab is often avoided in patients scheduled to undergo allogeneic SCT, based on data from a Japanese study that showed an increased risk of severe graft-versus-host disease (GVHD) in patients treated with mogamulizumab beforehand.[49] The relevance of these findings in other countries and the impact of different GVHD prophylaxis regimens in these patients remains to be determined.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Becker M, Hoppe RT, Knox SJ: Multiple courses of high-dose total skin electron beam therapy in the management of mycosis fungoides. Int J Radiat Oncol Biol Phys 32 (5): 1445-9, 1995. [PUBMED Abstract]
Trautinger F, Knobler R, Willemze R, et al.: EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome. Eur J Cancer 42 (8): 1014-30, 2006. [PUBMED Abstract]
Prince HM, Duvic M, Martin A, et al.: Phase III placebo-controlled trial of denileukin diftitox for patients with cutaneous T-cell lymphoma. J Clin Oncol 28 (11): 1870-7, 2010. [PUBMED Abstract]
Chinn DM, Chow S, Kim YH, et al.: Total skin electron beam therapy with or without adjuvant topical nitrogen mustard or nitrogen mustard alone as initial treatment of T2 and T3 mycosis fungoides. Int J Radiat Oncol Biol Phys 43 (5): 951-8, 1999. [PUBMED Abstract]
Quirós PA, Jones GW, Kacinski BM, et al.: Total skin electron beam therapy followed by adjuvant psoralen/ultraviolet-A light in the management of patients with T1 and T2 cutaneous T-cell lymphoma (mycosis fungoides). Int J Radiat Oncol Biol Phys 38 (5): 1027-35, 1997. [PUBMED Abstract]
Ysebaert L, Truc G, Dalac S, et al.: Ultimate results of radiation therapy for T1-T2 mycosis fungoides (including reirradiation). Int J Radiat Oncol Biol Phys 58 (4): 1128-34, 2004. [PUBMED Abstract]
Jones GW, Rosenthal D, Wilson LD: Total skin electron radiation for patients with erythrodermic cutaneous T-cell lymphoma (mycosis fungoides and the Sézary syndrome). Cancer 85 (9): 1985-95, 1999. [PUBMED Abstract]
Navi D, Riaz N, Levin YS, et al.: The Stanford University experience with conventional-dose, total skin electron-beam therapy in the treatment of generalized patch or plaque (T2) and tumor (T3) mycosis fungoides. Arch Dermatol 147 (5): 561-7, 2011. [PUBMED Abstract]
Micaily B, Miyamoto C, Kantor G, et al.: Radiotherapy for unilesional mycosis fungoides. Int J Radiat Oncol Biol Phys 42 (2): 361-4, 1998. [PUBMED Abstract]
Thomas TO, Agrawal P, Guitart J, et al.: Outcome of patients treated with a single-fraction dose of palliative radiation for cutaneous T-cell lymphoma. Int J Radiat Oncol Biol Phys 85 (3): 747-53, 2013. [PUBMED Abstract]
Herrmann JJ, Roenigk HH, Hurria A, et al.: Treatment of mycosis fungoides with photochemotherapy (PUVA): long-term follow-up. J Am Acad Dermatol 33 (2 Pt 1): 234-42, 1995. [PUBMED Abstract]
Ramsay DL, Lish KM, Yalowitz CB, et al.: Ultraviolet-B phototherapy for early-stage cutaneous T-cell lymphoma. Arch Dermatol 128 (7): 931-3, 1992. [PUBMED Abstract]
Querfeld C, Rosen ST, Kuzel TM, et al.: Long-term follow-up of patients with early-stage cutaneous T-cell lymphoma who achieved complete remission with psoralen plus UV-A monotherapy. Arch Dermatol 141 (3): 305-11, 2005. [PUBMED Abstract]
Kuzel TM, Roenigk HH, Samuelson E, et al.: Effectiveness of interferon alfa-2a combined with phototherapy for mycosis fungoides and the Sézary syndrome. J Clin Oncol 13 (1): 257-63, 1995. [PUBMED Abstract]
Olsen EA, Hodak E, Anderson T, et al.: Guidelines for phototherapy of mycosis fungoides and Sézary syndrome: A consensus statement of the United States Cutaneous Lymphoma Consortium. J Am Acad Dermatol 74 (1): 27-58, 2016. [PUBMED Abstract]
Phan K, Ramachandran V, Fassihi H, et al.: Comparison of Narrowband UV-B With Psoralen-UV-A Phototherapy for Patients With Early-Stage Mycosis Fungoides: A Systematic Review and Meta-analysis. JAMA Dermatol 155 (3): 335-341, 2019. [PUBMED Abstract]
Almohideb M, Walsh S, Walsh S, et al.: Bath Psoralen-ultraviolet A and Narrowband Ultraviolet B Phototherapy as Initial Therapy for Early-stage Mycosis Fungoides: A Retrospective Cohort of 267 Cases at the University of Toronto. Clin Lymphoma Myeloma Leuk 17 (9): 604-612, 2017. [PUBMED Abstract]
Elcin G, Duman N, Karahan S, et al.: Long-term follow-up of early mycosis fungoides patients treated with narrowband ultraviolet B phototherapy. J Dermatolog Treat 25 (3): 268-73, 2014. [PUBMED Abstract]
Edelson R, Berger C, Gasparro F, et al.: Treatment of cutaneous T-cell lymphoma by extracorporeal photochemotherapy. Preliminary results. N Engl J Med 316 (6): 297-303, 1987. [PUBMED Abstract]
Heald PW, Perez MI, McKiernan G, et al.: Extracorporeal photochemotherapy for CTCL. Prog Clin Biol Res 337: 443-7, 1990. [PUBMED Abstract]
Gao C, McCormack C, van der Weyden C, et al.: Prolonged survival with the early use of a novel extracorporeal photopheresis regimen in patients with Sézary syndrome. Blood 134 (16): 1346-1350, 2019. [PUBMED Abstract]
Lessin SR, Duvic M, Guitart J, et al.: Topical chemotherapy in cutaneous T-cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatol 149 (1): 25-32, 2013. [PUBMED Abstract]
de Quatrebarbes J, Estève E, Bagot M, et al.: Treatment of early-stage mycosis fungoides with twice-weekly applications of mechlorethamine and topical corticosteroids: a prospective study. Arch Dermatol 141 (9): 1117-20, 2005. [PUBMED Abstract]
Horwitz SM, Kim YH, Foss F, et al.: Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood 119 (18): 4115-22, 2012. [PUBMED Abstract]
Talpur R, Thompson A, Gangar P, et al.: Pralatrexate alone or in combination with bexarotene: long-term tolerability in relapsed/refractory mycosis fungoides. Clin Lymphoma Myeloma Leuk 14 (4): 297-304, 2014. [PUBMED Abstract]
Hughes CF, Khot A, McCormack C, et al.: Lack of durable disease control with chemotherapy for mycosis fungoides and Sézary syndrome: a comparative study of systemic therapy. Blood 125 (1): 71-81, 2015. [PUBMED Abstract]
Dummer R, Quaglino P, Becker JC, et al.: Prospective international multicenter phase II trial of intravenous pegylated liposomal doxorubicin monochemotherapy in patients with stage IIB, IVA, or IVB advanced mycosis fungoides: final results from EORTC 21012. J Clin Oncol 30 (33): 4091-7, 2012. [PUBMED Abstract]
Wollina U, Dummer R, Brockmeyer NH, et al.: Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T-cell lymphoma. Cancer 98 (5): 993-1001, 2003. [PUBMED Abstract]
Quereux G, Marques S, Nguyen JM, et al.: Prospective multicenter study of pegylated liposomal doxorubicin treatment in patients with advanced or refractory mycosis fungoides or Sézary syndrome. Arch Dermatol 144 (6): 727-33, 2008. [PUBMED Abstract]
Kaye FJ, Bunn PA, Steinberg SM, et al.: A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. N Engl J Med 321 (26): 1784-90, 1989. [PUBMED Abstract]
Rosen ST, Foss FM: Chemotherapy for mycosis fungoides and the Sézary syndrome. Hematol Oncol Clin North Am 9 (5): 1109-16, 1995. [PUBMED Abstract]
Zackheim HS, Epstein EH: Low-dose methotrexate for the Sézary syndrome. J Am Acad Dermatol 21 (4 Pt 1): 757-62, 1989. [PUBMED Abstract]
Miller VA, Benedetti FM, Rigas JR, et al.: Initial clinical trial of a selective retinoid X receptor ligand, LGD1069. J Clin Oncol 15 (2): 790-5, 1997. [PUBMED Abstract]
Duvic M, Hymes K, Heald P, et al.: Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol 19 (9): 2456-71, 2001. [PUBMED Abstract]
Querfeld C, Rosen ST, Guitart J, et al.: Results of an open-label multicenter phase 2 trial of lenalidomide monotherapy in refractory mycosis fungoides and Sézary syndrome. Blood 123 (8): 1159-66, 2014. [PUBMED Abstract]
Duvic M, Dummer R, Becker JC, et al.: Panobinostat activity in both bexarotene-exposed and -naïve patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur J Cancer 49 (2): 386-94, 2013. [PUBMED Abstract]
Olsen EA, Kim YH, Kuzel TM, et al.: Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25 (21): 3109-15, 2007. [PUBMED Abstract]
Piekarz RL, Frye R, Turner M, et al.: Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 27 (32): 5410-7, 2009. [PUBMED Abstract]
Foss FM, Ihde DC, Breneman DL, et al.: Phase II study of pentostatin and intermittent high-dose recombinant interferon alfa-2a in advanced mycosis fungoides/Sézary syndrome. J Clin Oncol 10 (12): 1907-13, 1992. [PUBMED Abstract]
Olsen EA, Bunn PA: Interferon in the treatment of cutaneous T-cell lymphoma. Hematol Oncol Clin North Am 9 (5): 1089-107, 1995. [PUBMED Abstract]
Molina A, Zain J, Arber DA, et al.: Durable clinical, cytogenetic, and molecular remissions after allogeneic hematopoietic cell transplantation for refractory Sezary syndrome and mycosis fungoides. J Clin Oncol 23 (25): 6163-71, 2005. [PUBMED Abstract]
Duvic M, Donato M, Dabaja B, et al.: Total skin electron beam and non-myeloablative allogeneic hematopoietic stem-cell transplantation in advanced mycosis fungoides and Sezary syndrome. J Clin Oncol 28 (14): 2365-72, 2010. [PUBMED Abstract]
Duarte RF, Boumendil A, Onida F, et al.: Long-term outcome of allogeneic hematopoietic cell transplantation for patients with mycosis fungoides and Sézary syndrome: a European society for blood and marrow transplantation lymphoma working party extended analysis. J Clin Oncol 32 (29): 3347-8, 2014. [PUBMED Abstract]
Schlaak M, Pickenhain J, Theurich S, et al.: Allogeneic stem cell transplantation versus conventional therapy for advanced primary cutaneous T-cell lymphoma. Cochrane Database Syst Rev 1: CD008908, 2012. [PUBMED Abstract]
Lechowicz MJ, Lazarus HM, Carreras J, et al.: Allogeneic hematopoietic cell transplantation for mycosis fungoides and Sezary syndrome. Bone Marrow Transplant 49 (11): 1360-5, 2014. [PUBMED Abstract]
Kim YH, Tavallaee M, Sundram U, et al.: Phase II Investigator-Initiated Study of Brentuximab Vedotin in Mycosis Fungoides and Sézary Syndrome With Variable CD30 Expression Level: A Multi-Institution Collaborative Project. J Clin Oncol 33 (32): 3750-8, 2015. [PUBMED Abstract]
Duvic M, Tetzlaff MT, Gangar P, et al.: Results of a Phase II Trial of Brentuximab Vedotin for CD30+ Cutaneous T-Cell Lymphoma and Lymphomatoid Papulosis. J Clin Oncol 33 (32): 3759-65, 2015. [PUBMED Abstract]
Kim YH, Bagot M, Pinter-Brown L, et al.: Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol 19 (9): 1192-1204, 2018. [PUBMED Abstract]
Treatment of Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous anaplastic large cell lymphoma presents in the skin only with no preexisting lymphoproliferative disease and no extracutaneous sites of involvement.[1–3] Patients with this type of lymphoma have disease ranging from clinically benign lymphomatoid papulosis, marked by localized nodules that may regress spontaneously, to progressive and systemic illness requiring aggressive doxorubicin-based combination chemotherapy. This spectrum has been called the primary cutaneous CD30-positive T-cell lymphoproliferative disorder.
Patients with localized disease usually undergo radiation therapy. With more disseminated involvement, watchful waiting or doxorubicin-based combination chemotherapy is used.[1–3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
de Bruin PC, Beljaards RC, van Heerde P, et al.: Differences in clinical behaviour and immunophenotype between primary cutaneous and primary nodal anaplastic large cell lymphoma of T-cell or null cell phenotype. Histopathology 23 (2): 127-35, 1993. [PUBMED Abstract]
Willemze R, Beljaards RC: Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders. A proposal for classification and guidelines for management and treatment. J Am Acad Dermatol 28 (6): 973-80, 1993. [PUBMED Abstract]
Kempf W, Pfaltz K, Vermeer MH, et al.: EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood 118 (15): 4024-35, 2011. [PUBMED Abstract]
Treatment of Subcutaneous Panniculitis-Like T-Cell Lymphoma
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is localized to subcutaneous tissue and can be associated with hemophagocytic lymphohistiocytosis (HLH).[1–4] Anecdotal reports suggest that the presence or absence of HLH is an important prognostic indicator.[5] Patients with SPTCL have cells that express alpha-beta phenotype.
Patients with gamma-delta phenotype have a more aggressive clinical course that is instead classified as primary cutaneous gamma-delta T-cell lymphoma.[6–8] For more information, see Peripheral T-Cell Non-Hodgkin Lymphoma Treatment.
The management of SPTCL depends on the clinical presentation—including the severity of symptoms, cytopenias, and the presence or absence of HLH—and the apparent disease trajectory and aggressiveness. Indolent or smoldering forms of SPTCL are often treated with immunosuppression, including oral methotrexate [9] or cyclosporine.[10] In contrast, aggressive forms are frequently treated with combination chemotherapy such as CHO(E)P (cyclophosphamide, doxorubicin, vincristine, and prednisone with or without etoposide) with variable responses per anecdotal reports.[11] The JAK2 inhibitor ruxolitinib has also been used in SPTCL with associated HLH, including anecdotal reports of responses in patients with disease that did not respond to CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) therapy.[12] In cases of particularly aggressive or relapsed disease, consolidation with allogeneic stem cell transplant has also been used.[13,14]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Go RS, Wester SM: Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer 101 (6): 1404-13, 2004. [PUBMED Abstract]
Marzano AV, Berti E, Paulli M, et al.: Cytophagic histiocytic panniculitis and subcutaneous panniculitis-like T-cell lymphoma: report of 7 cases. Arch Dermatol 136 (7): 889-96, 2000. [PUBMED Abstract]
Hoque SR, Child FJ, Whittaker SJ, et al.: Subcutaneous panniculitis-like T-cell lymphoma: a clinicopathological, immunophenotypic and molecular analysis of six patients. Br J Dermatol 148 (3): 516-25, 2003. [PUBMED Abstract]
Salhany KE, Macon WR, Choi JK, et al.: Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol 22 (7): 881-93, 1998. [PUBMED Abstract]
Willemze R: Cutaneous lymphomas with a panniculitic presentation. Semin Diagn Pathol 34 (1): 36-43, 2017. [PUBMED Abstract]
Massone C, Chott A, Metze D, et al.: Subcutaneous, blastic natural killer (NK), NK/T-cell, and other cytotoxic lymphomas of the skin: a morphologic, immunophenotypic, and molecular study of 50 patients. Am J Surg Pathol 28 (6): 719-35, 2004. [PUBMED Abstract]
Arnulf B, Copie-Bergman C, Delfau-Larue MH, et al.: Nonhepatosplenic gammadelta T-cell lymphoma: a subset of cytotoxic lymphomas with mucosal or skin localization. Blood 91 (5): 1723-31, 1998. [PUBMED Abstract]
Toro JR, Liewehr DJ, Pabby N, et al.: Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood 101 (9): 3407-12, 2003. [PUBMED Abstract]
Grinich E, Koon SM, Cascio MJ, et al.: Subcutaneous panniculitis-like T-cell lymphoma responsive to combination therapy with methotrexate and corticosteroids. Dermatol Online J 24 (9): , 2018. [PUBMED Abstract]
Willemze R, Jansen PM, Cerroni L, et al.: Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 111 (2): 838-45, 2008. [PUBMED Abstract]
Lévy R, Fusaro M, Guerin F, et al.: Efficacy of ruxolitinib in subcutaneous panniculitis-like T-cell lymphoma and hemophagocytic lymphohistiocytosis. Blood Adv 4 (7): 1383-1387, 2020. [PUBMED Abstract]
Weng W, Iragavarapu C, Weng GM, et al.: Long-term remission with allogeneic transplant in patients with refractory/relapsed cutaneous cytotoxic T-cell lymphoma. Blood Neoplasia 1 (2): 2024.
Ichii M, Hatanaka K, Imakita M, et al.: Successful treatment of refractory subcutaneous panniculitis-like T-cell lymphoma with allogeneic peripheral blood stem cell transplantation from HLA-mismatched sibling donor. Leuk Lymphoma 47 (10): 2250-2, 2006. [PUBMED Abstract]
Treatment of Primary Cutaneous Gamma-Delta T-Cell Lymphoma
Primary cutaneous gamma-delta T-cell lymphoma (PCGDTCL) is a rare and extremely aggressive form of cutaneous T-cell lymphoma with a poor prognosis. These patients may manifest involvement of the epidermis, dermis, subcutaneous region, or mucosa with or without ulceration. PCGDTCL is treated similarly to the most aggressive peripheral T-cell lymphomas, with CHO(E)P (cyclophosphamide doxorubicin, vincristine, and prednisone with or without etoposide).[1–5] For patients achieving remission, there are reports of prolonged survival following consolidation with allogeneic stem cell transplant.[6]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Arnulf B, Copie-Bergman C, Delfau-Larue MH, et al.: Nonhepatosplenic gammadelta T-cell lymphoma: a subset of cytotoxic lymphomas with mucosal or skin localization. Blood 91 (5): 1723-31, 1998. [PUBMED Abstract]
Toro JR, Liewehr DJ, Pabby N, et al.: Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood 101 (9): 3407-12, 2003. [PUBMED Abstract]
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008. [PUBMED Abstract]
Pro B, Allen P, Behdad A: Hepatosplenic T-cell lymphoma: a rare but challenging entity. Blood 136 (18): 2018-2026, 2020. [PUBMED Abstract]
Alberti-Violetti S, Maronese CA, Venegoni L, et al.: Primary Cutaneous Gamma-Delta T Cell Lymphomas: A Case Series and Overview of the Literature. Dermatopathology (Basel) 8 (4): 515-524, 2021. [PUBMED Abstract]
Isufi I, Seropian S, Gowda L, et al.: Outcomes for allogeneic stem cell transplantation in refractory mycosis fungoides and primary cutaneous gamma Delta T cell lymphomas. Leuk Lymphoma 61 (12): 2955-2961, 2020. [PUBMED Abstract]
Treatment of Primary Cutaneous Aggressive Epidermotropic CD8-Positive T-Cell Lymphoma
Primary cutaneous aggressive epidermotropic CD8-positive T-cell lymphoma is another rare and especially aggressive form of cutaneous T-cell lymphoma. Patients typically present with ulcerative plaques or tumors, and mucosal involvement is common. Neoplastic cells are characterized by expression of CD8 and a cytotoxic phenotype.[1] Multimodal chemotherapy, such as CHO(E)P (cyclophosphamide, doxorubicin, vincristine, and prednisone with or without etoposide), is often used. Outcomes are generally poor, and prolonged remissions are extremely uncommon in the absence of allogeneic stem cell transplant.[2]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Nofal A, Abdel-Mawla MY, Assaf M, et al.: Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: proposed diagnostic criteria and therapeutic evaluation. J Am Acad Dermatol 67 (4): 748-59, 2012. [PUBMED Abstract]
Guitart J, Martinez-Escala ME, Subtil A, et al.: Primary cutaneous aggressive epidermotropic cytotoxic T-cell lymphomas: reappraisal of a provisional entity in the 2016 WHO classification of cutaneous lymphomas. Mod Pathol 30 (5): 761-772, 2017. [PUBMED Abstract]
Key References for Mycosis Fungoides and Other Cutaneous T-Cell Lymphomas
These references have been identified by members of the PDQ Adult Treatment Editorial Board as significant in the field of mycosis fungoides and other cutaneous T-cell lymphoma treatment. This list is provided to inform users of important studies that have helped shape the current understanding of and treatment options for MF/SS. Listed after each reference are the sections within this summary where the reference is cited.
Agar NS, Wedgeworth E, Crichton S, et al.: Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol 28 (31): 4730-9, 2010. [PUBMED Abstract]
Hughes CF, Khot A, McCormack C, et al.: Lack of durable disease control with chemotherapy for mycosis fungoides and Sézary syndrome: a comparative study of systemic therapy. Blood 125 (1): 71-81, 2015. [PUBMED Abstract]
Kadin ME, Hughey LC, Wood GS: Large-cell transformation of mycosis fungoides-differential diagnosis with implications for clinical management: a consensus statement of the US Cutaneous Lymphoma Consortium. J Am Acad Dermatol 70 (2): 374-6, 2014. [PUBMED Abstract]
Quaglino P, Pimpinelli N, Berti E, et al.: Time course, clinical pathways, and long-term hazards risk trends of disease progression in patients with classic mycosis fungoides: a multicenter, retrospective follow-up study from the Italian Group of Cutaneous Lymphomas. Cancer 118 (23): 5830-9, 2012. [PUBMED Abstract]
Talpur R, Singh L, Daulat S, et al.: Long-term outcomes of 1,263 patients with mycosis fungoides and Sézary syndrome from 1982 to 2009. Clin Cancer Res 18 (18): 5051-60, 2012. [PUBMED Abstract]
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Added text to state that established criteria for defining Sézary syndrome generally require identification of (1) a circulating T-cell clone by rearrangement of the T-cell receptor, plus (2) a level of Sézary cells in the blood of at least 1,000/μL (cited 2007 Olsen et al. as reference 3 and 2022 Olsen et al. as reference 4). It is less clear if other findings are reliable staging data in the absence of blood Sézary cell levels of at least 1,000/μL (cited Chrisman et al. as reference 5).
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of mycosis fungoides and other cutaneous T-cell lymphomas. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Mycosis Fungoides and Other Cutaneous T-Cell Lymphomas Treatment are:
Eric J. Seifter, MD (Johns Hopkins University)
Cole H. Sterling, MD (Johns Hopkins University)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Mycosis Fungoides and Other Cutaneous T-Cell Lymphomas Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/lymphoma/hp/mycosis-fungoides-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389288]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Primary central nervous system (CNS) lymphoma is lymphoma limited to the cranial-spinal axis, including the brain, spinal cord, cerebrospinal fluid (leptomeningeal space), and vitreoretinal space (ocular space), without systemic disease (stage IE).[1] This disease has increasingly been seen among immunocompromised patients, such as those with HIV. Immunosuppression-related primary CNS lymphomas are almost always associated with the Epstein-Barr virus.
Histology
Almost all primary CNS lymphomas are diffuse large B-cell lymphomas of the activated B-cell nongerminal center subtype with additional pathogenic variants in the B-cell receptor signaling pathway, especially MYD88 and CD79B variants.[1] However, patients with immunosuppression-related disease almost never have an activated B-cell phenotype.[2]
More than 95% of patients with primary CNS lymphoma have B-cell phenoptype. However, in a retrospective series with data collected from 12 cancer centers, the 45 patients with CNS lymphoma of T-cell phenotype showed no difference in presentation or outcome.[3]
Anecdotal cases of primary CNS Hodgkin lymphoma have also been reported.[4]
Diagnostic Evaluation
Computed tomography (CT) scans are used to diagnose primary CNS lymphoma. These scans may show ring enhancement in 50% of patients with HIV, while homogenous enhancement is almost always seen in patients without HIV.[5]
Positron emission tomography (PET)–CT scans are used to exclude occult systemic disease of the chest, abdomen, and pelvis. A bone marrow biopsy may be excluded with a clear PET-CT scan.[6] All compartments of the CNS should be evaluated, even if asymptomatic, including the vitreoretinal compartment and the cerebrospinal fluid (CSF) when feasible.
In one prospective case series of 282 patients with primary CNS lymphoma, 17% of patients were found to have meningeal dissemination by cytomorphology, polymerase chain reaction of rearranged immunoglobulin heavy-chain genes, or meningeal enhancement on magnetic resonance imaging.[7]
Prognosis and Prognostic Factors
Primary CNS lymphoma is usually aggressive, and the median overall survival in published trials generally ranges from 2 to 5 years.[8,9] However, a retrospective case series of 40 patients with low-grade primary CNS lymphoma, derived from 18 cancer centers in five countries, reported a better long-term outcome (median survival, 7 years).[10][Level of evidence C3]
Poor prognostic factors for primary CNS lymphoma include:[11]
Age older than 60 years.
HIV positivity.
Elevated serum lactate dehydrogenase level.
Elevated CSF protein concentration.
Involvement of nonhemispheric areas of the brain (periventricular, basal ganglia, brainstem, and cerebellum).
Intraocular disease and concomitant brain involvement.[12] When tumor progression occurs, it is usually confined to the CNS and/or the eye.[1]
Older age and HIV positivity are the most clinically relevant poor prognostic factors. However, the prognosis for patients with HIV-associated primary CNS lymphoma has improved with the use of highly active antiretroviral therapy.[13] These patients are treated with the same paradigm as patients who do not have HIV or immunosuppression.
References
Schaff LR, Grommes C: Primary central nervous system lymphoma. Blood 140 (9): 971-979, 2022. [PUBMED Abstract]
Gandhi MK, Hoang T, Law SC, et al.: EBV-associated primary CNS lymphoma occurring after immunosuppression is a distinct immunobiological entity. Blood 137 (11): 1468-1477, 2021. [PUBMED Abstract]
Shenkier TN, Blay JY, O’Neill BP, et al.: Primary CNS lymphoma of T-cell origin: a descriptive analysis from the international primary CNS lymphoma collaborative group. J Clin Oncol 23 (10): 2233-9, 2005. [PUBMED Abstract]
Gerstner ER, Abrey LE, Schiff D, et al.: CNS Hodgkin lymphoma. Blood 112 (5): 1658-61, 2008. [PUBMED Abstract]
Fine HA, Mayer RJ: Primary central nervous system lymphoma. Ann Intern Med 119 (11): 1093-104, 1993. [PUBMED Abstract]
Jelicic J, Hansen DL, Carlsen SS, et al.: Is it possible to omit bone marrow biopsy in diagnostic workup in patients with newly diagnosed primary CNS lymphoma? A retrospective analysis in the PET/CT era. [Abstract] Blood 140 (Suppl 1): A-627, 1334-5, 2022.
Fischer L, Martus P, Weller M, et al.: Meningeal dissemination in primary CNS lymphoma: prospective evaluation of 282 patients. Neurology 71 (14): 1102-8, 2008. [PUBMED Abstract]
Fox CP, Phillips EH, Smith J, et al.: Guidelines for the diagnosis and management of primary central nervous system diffuse large B-cell lymphoma. Br J Haematol 184 (3): 348-363, 2019. [PUBMED Abstract]
Jahnke K, Korfel A, O’Neill BP, et al.: International study on low-grade primary central nervous system lymphoma. Ann Neurol 59 (5): 755-62, 2006. [PUBMED Abstract]
Lukas RV, Stupp R, Gondi V, et al.: Primary Central Nervous System Lymphoma-PART 1: Epidemiology, Diagnosis, Staging, and Prognosis. Oncology (Williston Park) 32 (1): 17-22, 2018. [PUBMED Abstract]
Kreher S, Strehlow F, Martus P, et al.: Prognostic impact of intraocular involvement in primary CNS lymphoma: experience from the G-PCNSL-SG1 trial. Ann Hematol 94 (3): 409-14, 2015. [PUBMED Abstract]
Gupta NK, Nolan A, Omuro A, et al.: Long-term survival in AIDS-related primary central nervous system lymphoma. Neuro Oncol 19 (1): 99-108, 2017. [PUBMED Abstract]
Treatment of Primary Central Nervous System Lymphoma
Treatment Options for Primary Central Nervous System (CNS) Lymphoma
Treatment options for primary CNS lymphoma include:
Trials using chemotherapy alone were justified because of the unsatisfactory results of using whole-brain radiation therapy (WBRT) alone [1,2] and significant neurological toxicity using high-dose methotrexate or other chemotherapeutic agents that cross the blood-brain barrier in combination with WBRT.[3–5]
Severe, delayed, neurological toxic effects were rarely seen in chemotherapy-only trials in the absence of subsequent radiation therapy. However, salvage radiation can be given for relapsed or refractory disease, sometimes at reduced dosage.[6,7]
Numerous phase I and phase II studies over two decades established the following active drugs for induction therapy or for treatment of relapsing disease. The following drugs have been used as single agents and in combinations:
High-dose methotrexate.[8–13] Outside of clinical trials, high-dose methotrexate is the most frequently used standard induction therapy.[14] However, it is given to inpatients, and it is too toxic for patients with a creatinine clearance under 35 cc/min or for most patients older than 75 years.
Lenalidomide with rituximab.[15] When high-dose chemotherapy is not feasible, this is the most frequently used combination. Lenalidomide can be initiated at low doses (e.g., 5 mg daily, 21 out of 28 days) in patients with renal insufficiency.
Evidence (chemotherapy with or without other therapy):
The International Extranodal Lymphoma Study Group evaluated three different induction combinations in 227 patients with newly diagnosed, HIV-negative primary CNS lymphoma. Patients were randomly assigned to receive high-dose methotrexate (HD MTX) + high-dose cytarabine (HDA) (group 1), HD MTX + HDA + rituximab (R) (group 2), or HD MTX + HDA + R + thiotepa (the MATRix regimen) (group 3).[26][Level of evidence A1]
With a median follow-up of 88 months, the overall survival (OS) rate was 21% for group 1, 37% for group 2, and 56% for group 3.
The groups were compared as follows:
Group 1 versus group 2: hazard ratio (HR), 0.64; 95% confidence interval (CI), 0.41–0.99; P = .04.
Group 1 versus group 3: HR, 0.42; 95% CI, 0.24–0.64; P = .00005.
Group 2 versus group 3: HR, 0.66; 95% CI, 0.44–0.98; P = .04.
The OS rate favored the complete MATRix regimen in all comparisons.[26][Level of evidence A1]
The 113 patients who attained a complete response, partial response, or stable disease were randomly assigned to 36 Gy whole-brain radiation therapy versus autologous stem cell transplant (SCT) after thiotepa + bic-chloroethylnitrosourea + carmustine (BCNU) conditioning. No significant differences were seen in 7-year progression-free survival (PFS) or OS in regard to consolidation in this phase II trial.[26][Level of evidence B1]
The thiotepa + BCNU conditioning regimen was well tolerated by patients aged 60 to 70 years. It had improved tolerability compared with prior reports using the more intensive thiotepa + busulfan regimen.[26][Level of evidence D]
Patients who received the two-drug combination had a complete remission rate of 23% (interquartile range [IQR], 14%‒31%; HR, 0.46; 95% CI, 0.28‒0.74). Patients who received the three-drug combination had a complete remission rate of 30% (IQR, 21%‒42%; HR, 0.61; 95% CI, 0.40‒0.94). Patients who received the four-drug MATRix combination had a complete remission rate of 49% (95% CI, 38%‒60%).
The addition of rituximab and thiotepa to high-dose methotrexate plus cytarabine resulted in a significant improvement in complete response, PFS, and OS.[26][Level of evidence A1]
In a randomized, nonblinded multicenter trial, 79 patients were assigned to receive high-dose methotrexate with or without cytarabine.[27][Level of evidence B1]
The 3-year PFS rate was better for patients who received the two-drug regimen (HR, 0.54; 95% CI, 0.31–0.92; P = .01).
There was no statistical difference in the 3-year OS rate (46% for the two-drug regimen vs. 32% for the one-drug regimen; HR, 0.65; 95% CI, 0.38–1.13; P = .07).
This trial was the basis for setting the combination of high-dose methotrexate and high-dose cytarabine as the control arm in the MATRix trial.
In a randomized, prospective, multicenter trial, 200 patients were assigned to receive intravenous high-dose methotrexate, carmustine, teniposide, and oral prednisone with or without rituximab.[28][Level of evidence B1]
With a median follow-up of 32.9 months, there was no difference in the 1-year event-free survival (EFS) rate: 52% with rituximab (95% CI, 42%−61%) and 49% without rituximab (95% CI, 39%−58%; HR, 1.00; 95% CI, 0.70−1.43; P = .99).
Several other combination induction regimens were created empirically and presented as phase II trials.[29][Level of evidence C3]
High-dose methotrexate regimens delivered with rituximab and other chemotherapeutic agents is used for induction therapy. The MATRix regimen described above has become one such standard based on randomized OS benefit with a four-drug regimen versus a two-drug or a three-drug regimen.[26][Level of evidence 1A] The MATRix regimen has never been compared with some of the other combination therapies mentioned above. A meta-analysis of rituximab randomized trials found improved PFS with the addition of rituximab (HR, 0.65; 95% CI, 0.45–0.95) but no difference in OS.[30][Level of evidence B2]
Consolidation after induction chemotherapy
Consolidation therapy with or without WBRT
Evidence (consolidation therapy with or without WBRT):
In a prospective, randomized, phase II trial, 87 patients (median age, 59–66 years) had induction therapy with high-dose methotrexate + high-dose cytarabine + rituximab + procarbazine + vincristine. Afterward, 37 patients had consolidation therapy with low-dose WBRT at 23.4 Gy.[31]
With a median follow-up of 55 months, the 2-year intention-to-treat PFS was 78% with low-dose WBRT versus 54% without low-dose WBRT (HR, 0.51; P = .015).[31][Level of evidence B1]
Investigator-assessed neurotoxicity was less than 15% in each arm and not significantly different. Another small phase II trial that examined low-dose WBRT at 23.4 Gy also found no increased problems seen on neuropsychological testing.[32,33]
In a prospective, randomized, phase II trial of 97 newly diagnosed patients who received high-dose methotrexate combination therapy for induction, patients were randomly assigned to receive WBRT at 40 Gy versus autologous SCT.[34]
With a median follow-up of 8 years, deterioration of balance was significantly greater for patients who received WBRT versus patients who received autologous SCT (52% vs. 10%, P ≤ .001). Worsening neurocognition was also greater for patients who received WBRT (64% vs. 13%, P < .001).
The 8-year EFS rate was 67% for patients who received autologous SCT versus 39% for patients who received WBRT (HR, 0.13; P < .001). There was no significant difference in OS (69% for autologous SCT vs. 54% for WBRT).[34][Level of evidence B1]
The authors concluded that 40 Gy WBRT should be avoided in first-line treatment because of its neurotoxicity and inferior efficacy.
In a prospective, randomized trial of 551 immunocompetent patients with newly diagnosed primary CNS lymphoma, all patients received induction chemotherapy with six cycles of high-dose methotrexate (4 g/m2) with or without ifosfamide. After chemotherapy was completed, responders were randomly assigned to receive either WBRT (45 Gy) or no treatment for complete-response patients and cytarabine for partial-response patients.[35]
There was no statistical difference in median OS at 32.4 months for patients who received WBRT versus at 37.1 months for those who did not receive WBRT (HR, 1.06; 95% CI, 0.80–1.40; P = .71).[35][Level of evidence A1]
Treatment-related neurotoxic effects were significantly worse in the WBRT arm.
In a prospective, randomized trial, 410 immunocompetent patients with newly diagnosed primary CNS lymphoma were scheduled to receive high-dose methotrexate. Patients were randomly assigned to receive either WBRT or no radiation therapy.[36]
In the intent-to-treat population, WBRT was associated with longer PFS at 15.4 months versus 9.9 months (HR, 0.79; 95% CI, 0.64–0.98; P = .034). There was no difference in OS at 32.4 months versus 36.1 months (HR, 0.98; 95% CI, 0.79–1.26; P = .98).[36][Level of evidence B1]
Long-term treatment-related neurotoxic effects were not reported, and the induction chemotherapy would now be considered substandard.
Summary
The significant neurotoxicity of standard-dose WBRT [37] has reduced its role to short-duration disease control at relapse, when the expected survival is short enough that the benefit outweighs the longer-term neurological consequences. For patients unable to undergo consolidation with autologous SCT due to age, performance status, or comorbidities, low-dose WBRT (23.4 Gy) would be a consolidation option.
Consolidation therapy with or without autologous SCT
Evidence (consolidation therapy with or without autologous SCT):
In a prospective, randomized trial published in abstract form, 346 patients with newly diagnosed primary CNS lymphoma (patients aged 65 years and younger and patients aged 66–70 years with a performance status of 2 or lower) underwent induction therapy with 4 cycles of rituximab, high-dose methotrexate, high-dose cytarabine, and thiotepa (IELSG43 [NCT02531841]). Eighty-six patients discontinued treatment due to toxicity or progression. Of the 260 patients who completed induction therapy, 229 patients were randomly assigned to receive either chemotherapy consolidation with rituximab, dexamethasone, etoposide, ifosfamide, and carboplatin (n = 115) or autologous SCT using BCNU and thiotepa (n = 114).[38][Level of evidence A1]
With a median follow-up of 44 months, the 3-year OS rate was 86% (78%–91%) for autologous SCT versus 71% (61%–78%) for chemotherapy alone (HR, 0.47; P = .01).[38][Level of evidence A1]
The 3-year PFS rate was 79% (71%–86%) for autologous SCT versus 53% (43%–62%) for chemotherapy alone.
There was no negative effect on neurocognitive function in either arm in the absence of progression.
This was the first randomized trial establishing a survival advantage for newly diagnosed patients receiving autologous SCT who are young enough and healthy enough to endure this aggressive approach.
In a prospective, randomized, phase II trial, 97 newly diagnosed patients receiving high-dose methotrexate combination therapy for induction were randomly assigned to receive WBRT at 40 Gy or autologous SCT.[34]
With a median follow-up of 8 years, deterioration of balance was significantly greater for patients who received WBRT than for patients who received autologous SCT (52% vs. 10%, P ≤ .001). Worsening neurocognition was also greater for patients who received WBRT (64% vs. 13%, P < .001).
The 8-year EFS rate was 67% for patients who received autologous SCT versus 39% for patients who received WBRT (HR, 0.13; P < .001). There was no difference in OS (69% for autologous SCT vs. 65% for WBRT).[34][Level of evidence B1]
The authors concluded that 40 Gy WBRT should be avoided in first-line treatment because of its neurotoxicity and inferior efficacy.
A prospective, randomized, phase II trial included newly diagnosed patients aged 75 years and younger. Patients received intensive induction therapy with high-dose methotrexate at 8 g/m2 every 2 weeks, temozolomide, rituximab, and high-dose cytarabine. Patients were randomly assigned at the start of therapy to consolidation with thiotepa + BCNU conditioning with autologous SCT or nonmyeloablative therapy with high-dose cytarabine with a 46-hour infusion of etoposide (EA).[39]
With a median follow-up of 3.8 years, the median PFS was 6 years (95% CI, 3.9–not reached [NR]) for autologous SCT versus 2.4 years (95% CI, 0.6–NR) for EA (P = .02).[39][Level of evidence B1]
PFS from the time of consolidation was not statistically different (HR, 0.58; 95% CI, 0.25–1.36; P = .21).
There was no statistically significant difference in the 3-year OS rate: 83% (69%–91%) for autologous SCT and 72% (57%–82%) for EA.
This trial showed the limitations of induction therapy, which affected the impact of consolidation therapy.
Several phase II studies evaluated consolidation with intensive chemotherapy supported by autologous SCT.[19,20,23,40–43] This approach is appropriate for patients aged 80 years and under with few comorbidities, good performance status, and an adequate response to induction therapy.
Summary
Consolidation therapy with autologous SCT results in an OS advantage for newly diagnosed patients with good performance status, few comorbidities, and an adequate response to induction therapy (in this case, the MATRix regimen: high-dose methotrexate, high-dose cytarabine, rituximab, and temozolomide).[39] For patients unable to proceed to autologous SCT, consolidation with low-dose WBRT (32.4 Gy) or nonmyeloablative therapy might be considered.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Pollack IF, Lunsford LD, Flickinger JC, et al.: Prognostic factors in the diagnosis and treatment of primary central nervous system lymphoma. Cancer 63 (5): 939-47, 1989. [PUBMED Abstract]
Nelson DF, Martz KL, Bonner H, et al.: Non-Hodgkin’s lymphoma of the brain: can high dose, large volume radiation therapy improve survival? Report on a prospective trial by the Radiation Therapy Oncology Group (RTOG): RTOG 8315. Int J Radiat Oncol Biol Phys 23 (1): 9-17, 1992. [PUBMED Abstract]
Fisher B, Seiferheld W, Schultz C, et al.: Secondary analysis of Radiation Therapy Oncology Group study (RTOG) 9310: an intergroup phase II combined modality treatment of primary central nervous system lymphoma. J Neurooncol 74 (2): 201-5, 2005. [PUBMED Abstract]
Ekenel M, Iwamoto FM, Ben-Porat LS, et al.: Primary central nervous system lymphoma: the role of consolidation treatment after a complete response to high-dose methotrexate-based chemotherapy. Cancer 113 (5): 1025-31, 2008. [PUBMED Abstract]
van der Meulen M, Dirven L, Habets EJJ, et al.: Cognitive functioning and health-related quality of life in patients with newly diagnosed primary CNS lymphoma: a systematic review. Lancet Oncol 19 (8): e407-e418, 2018. [PUBMED Abstract]
Khimani NB, Ng AK, Chen YH, et al.: Salvage radiotherapy in patients with recurrent or refractory primary or secondary central nervous system lymphoma after methotrexate-based chemotherapy. Ann Oncol 22 (4): 979-84, 2011. [PUBMED Abstract]
Shah GD, Yahalom J, Correa DD, et al.: Combined immunochemotherapy with reduced whole-brain radiotherapy for newly diagnosed primary CNS lymphoma. J Clin Oncol 25 (30): 4730-5, 2007. [PUBMED Abstract]
Gavrilovic IT, Hormigo A, Yahalom J, et al.: Long-term follow-up of high-dose methotrexate-based therapy with and without whole brain irradiation for newly diagnosed primary CNS lymphoma. J Clin Oncol 24 (28): 4570-4, 2006. [PUBMED Abstract]
Blay JY, Conroy T, Chevreau C, et al.: High-dose methotrexate for the treatment of primary cerebral lymphomas: analysis of survival and late neurologic toxicity in a retrospective series. J Clin Oncol 16 (3): 864-71, 1998. [PUBMED Abstract]
Batchelor T, Carson K, O’Neill A, et al.: Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96-07. J Clin Oncol 21 (6): 1044-9, 2003. [PUBMED Abstract]
Hoang-Xuan K, Taillandier L, Chinot O, et al.: Chemotherapy alone as initial treatment for primary CNS lymphoma in patients older than 60 years: a multicenter phase II study (26952) of the European Organization for Research and Treatment of Cancer Brain Tumor Group. J Clin Oncol 21 (14): 2726-31, 2003. [PUBMED Abstract]
Pels H, Schmidt-Wolf IG, Glasmacher A, et al.: Primary central nervous system lymphoma: results of a pilot and phase II study of systemic and intraventricular chemotherapy with deferred radiotherapy. J Clin Oncol 21 (24): 4489-95, 2003. [PUBMED Abstract]
Juergens A, Pels H, Rogowski S, et al.: Long-term survival with favorable cognitive outcome after chemotherapy in primary central nervous system lymphoma. Ann Neurol 67 (2): 182-9, 2010. [PUBMED Abstract]
Schaff LR, Grommes C: Primary central nervous system lymphoma. Blood 140 (9): 971-979, 2022. [PUBMED Abstract]
Ghesquieres H, Chevrier M, Laadhari M, et al.: Lenalidomide in combination with intravenous rituximab (REVRI) in relapsed/refractory primary CNS lymphoma or primary intraocular lymphoma: a multicenter prospective ‘proof of concept’ phase II study of the French Oculo-Cerebral lymphoma (LOC) Network and the Lymphoma Study Association (LYSA)†. Ann Oncol 30 (4): 621-628, 2019. [PUBMED Abstract]
Chen YB, Batchelor T, Li S, et al.: Phase 2 trial of high-dose rituximab with high-dose cytarabine mobilization therapy and high-dose thiotepa, busulfan, and cyclophosphamide autologous stem cell transplantation in patients with central nervous system involvement by non-Hodgkin lymphoma. Cancer 121 (2): 226-33, 2015. [PUBMED Abstract]
Mocikova H, Pytlik R, Sykorova A, et al.: Role of rituximab in treatment of patients with primary central nervous system lymphoma: a retrospective analysis of the Czech lymphoma study group registry. Leuk Lymphoma 57 (12): 2777-2783, 2016. [PUBMED Abstract]
Ferreri AJ, Cwynarski K, Pulczynski E, et al.: Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: results of the first randomisation of the International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2 trial. Lancet Haematol 3 (5): e217-27, 2016. [PUBMED Abstract]
Schorb E, Fox CP, Fritsch K, et al.: High-dose thiotepa-based chemotherapy with autologous stem cell support in elderly patients with primary central nervous system lymphoma: a European retrospective study. Bone Marrow Transplant 52 (8): 1113-1119, 2017. [PUBMED Abstract]
Rubenstein JL, Hsi ED, Johnson JL, et al.: Intensive chemotherapy and immunotherapy in patients with newly diagnosed primary CNS lymphoma: CALGB 50202 (Alliance 50202). J Clin Oncol 31 (25): 3061-8, 2013. [PUBMED Abstract]
Illerhaus G, Schorb E, Kasenda B: Novel agents for primary central nervous system lymphoma: evidence and perspectives. Blood 132 (7): 681-688, 2018. [PUBMED Abstract]
Grommes C, Tang SS, Wolfe J, et al.: Phase 1b trial of an ibrutinib-based combination therapy in recurrent/refractory CNS lymphoma. Blood 133 (5): 436-445, 2019. [PUBMED Abstract]
Omuro A, Correa DD, DeAngelis LM, et al.: R-MPV followed by high-dose chemotherapy with TBC and autologous stem-cell transplant for newly diagnosed primary CNS lymphoma. Blood 125 (9): 1403-10, 2015. [PUBMED Abstract]
Tun HW, Johnston PB, DeAngelis LM, et al.: Phase 1 study of pomalidomide and dexamethasone for relapsed/refractory primary CNS or vitreoretinal lymphoma. Blood 132 (21): 2240-2248, 2018. [PUBMED Abstract]
Nayak L, Iwamoto FM, LaCasce A, et al.: PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 129 (23): 3071-3073, 2017. [PUBMED Abstract]
Ferreri AJM, Cwynarski K, Pulczynski E, et al.: Long-term efficacy, safety and neurotolerability of MATRix regimen followed by autologous transplant in primary CNS lymphoma: 7-year results of the IELSG32 randomized trial. Leukemia 36 (7): 1870-1878, 2022. [PUBMED Abstract]
Ferreri AJ, Reni M, Foppoli M, et al.: High-dose cytarabine plus high-dose methotrexate versus high-dose methotrexate alone in patients with primary CNS lymphoma: a randomised phase 2 trial. Lancet 374 (9700): 1512-20, 2009. [PUBMED Abstract]
Bromberg JEC, Issa S, Bakunina K, et al.: Rituximab in patients with primary CNS lymphoma (HOVON 105/ALLG NHL 24): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 20 (2): 216-228, 2019. [PUBMED Abstract]
Houillier C, Soussain C, Ghesquières H, et al.: Management and outcome of primary CNS lymphoma in the modern era: An LOC network study. Neurology 94 (10): e1027-e1039, 2020. [PUBMED Abstract]
Schmitt AM, Herbrand AK, Fox CP, et al.: Rituximab in primary central nervous system lymphoma-A systematic review and meta-analysis. Hematol Oncol 37 (5): 548-557, 2019. [PUBMED Abstract]
Omuro AM, DeAngelis LM, Karrison T, et al.: Randomized phase II study of rituximab, methotrexate (MTX), procarbazine, vincristine, and cytarabine (R-MPV-A) with and without low-dose whole-brain radiotherapy (LD-WBRT) for newly diagnosed primary CNS lymphoma (PCNSL). [Abstract] J Clin Oncol 38 (Suppl 15): A-2501, 2020.
Morris PG, Correa DD, Yahalom J, et al.: Rituximab, methotrexate, procarbazine, and vincristine followed by consolidation reduced-dose whole-brain radiotherapy and cytarabine in newly diagnosed primary CNS lymphoma: final results and long-term outcome. J Clin Oncol 31 (31): 3971-9, 2013. [PUBMED Abstract]
Lesueur P, Damaj G, Hoang-Xuan K, et al.: Reduced-dose WBRT as consolidation treatment for patients with primary CNS lymphoma: an LOC network study. Blood Adv 6 (16): 4807-4815, 2022. [PUBMED Abstract]
Houillier C, Dureau S, Taillandier L, et al.: Radiotherapy or Autologous Stem-Cell Transplantation for Primary CNS Lymphoma in Patients Age 60 Years and Younger: Long-Term Results of the Randomized Phase II PRECIS Study. J Clin Oncol 40 (32): 3692-3698, 2022. [PUBMED Abstract]
Thiel E, Korfel A, Martus P, et al.: High-dose methotrexate with or without whole brain radiotherapy for primary CNS lymphoma (G-PCNSL-SG-1): a phase 3, randomised, non-inferiority trial. Lancet Oncol 11 (11): 1036-47, 2010. [PUBMED Abstract]
Korfel A, Thiel E, Martus P, et al.: Randomized phase III study of whole-brain radiotherapy for primary CNS lymphoma. Neurology 84 (12): 1242-8, 2015. [PUBMED Abstract]
Illerhaus G, Ferreri AJ, Binder M, et al.: Effects on Survival of Non-Myeloablative Chemoimmunotherapy Compared to High-Dose Chemotherapy Followed By Autologous Stem Cell Transplantation (HDC-ASCT) As Consolidation Therapy in Patients with Primary CNS Lymphoma – Results of an International Randomized Phase III Trial (MATRix/IELSG43). [Abstract] Blood 140 (Suppl 2): A-LBA-3, 2022.
Batchelor T, Giri S, Ruppert A, et al.: Myeloablative versus non-myeloablative consolidative chemotherapy for newly diagnosed primary central nervous system lymphoma: Results of CALGB 51101 (Alliance). [Abstract] J Clin Oncol 39 (Suppl 15): A-7506, 2021.
Illerhaus G, Kasenda B, Ihorst G, et al.: High-dose chemotherapy with autologous haemopoietic stem cell transplantation for newly diagnosed primary CNS lymphoma: a prospective, single-arm, phase 2 trial. Lancet Haematol 3 (8): e388-97, 2016. [PUBMED Abstract]
Kasenda B, Schorb E, Fritsch K, et al.: Prognosis after high-dose chemotherapy followed by autologous stem-cell transplantation as first-line treatment in primary CNS lymphoma–a long-term follow-up study. Ann Oncol 23 (10): 2670-5, 2012. [PUBMED Abstract]
Ferreri AJ, Illerhaus G: The role of autologous stem cell transplantation in primary central nervous system lymphoma. Blood 127 (13): 1642-9, 2016. [PUBMED Abstract]
DeFilipp Z, Li S, El-Jawahri A, et al.: High-dose chemotherapy with thiotepa, busulfan, and cyclophosphamide and autologous stem cell transplantation for patients with primary central nervous system lymphoma in first complete remission. Cancer 123 (16): 3073-3079, 2017. [PUBMED Abstract]
Treatment of Recurrent Primary Central Nervous System Lymphoma
Treatment Options for Recurrent Primary Central Nervous System (CNS) Lymphoma
The prognosis for patients with recurrent CNS lymphoma is poor, with a median survival of 6 to 12 months, but up to 43 to 50 months if autologous stem cell transplant (SCT) consolidation is performed (if not applied previously).[1] This finding implies that deferred autologous SCT until first relapse may still result in longer-term survival. The prognosis is worse for patients 60 years and older, who account for more than 50% of cases.[2]
Treatment options for recurrent primary CNS lymphoma include:
Patients with recurrence after high-dose methotrexate-based combination chemotherapy may try autologous SCT consolidation after reinduction of remission with single-agent or combination therapy from the following options:[3]
Patients deemed ineligible for transplant can receive palliative care with these agents.
Evidence (reinduction therapy):
A phase I/II clinical trial evaluated CD19-directed CAR T-cell therapy using tisagenlecleucel in patients with relapsed primary CNS lymphoma.[8][Level of evidence C3]
One-half of the patients (6 of 12) had a complete response.
Three patients maintained a complete response at 9 months, 12 months, and 23 months at the time of data cutoff.
Five patients had low-grade immune cell–assisted neurotoxicity.
One patient had grade 3 neurotoxicity.
CAR T-cell therapy provides an option for patients with relapsed primary CNS lymphoma.[8]
The DA-TEDDI-R regimen incorporates temozolomide, etoposide, liposomal doxorubicin, dexamethasone, ibrutinib, and rituximab.[7][Level of evidence C3]
Among 18 patients who received this regimen (five previously untreated), the complete remission rate was 86%, but high rates (39%) of invasive aspergillosis were reported.
Further studies of this regimen are under way (NCT03964090 and NCT02203526). Dexamethasone should be avoided with ibrutinib single agent or combination therapy due to the risk of serious fungal infections. This approach requires access to intravenous antifungal agents not available outside of a clinical trial. By eliminating dexamethasone and ibrutinib, the other drugs may be used together with less risk of fungal infections.
In a phase II study, patients with relapsed or refractory primary CNS lymphoma were treated with rituximab plus lenalidomide.[4][Level of evidence C3]
An overall response rate of 36% was reported.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Schenone L, Houillier C, Tanguy ML, et al.: Intensive chemotherapy followed by autologous stem cell transplantation in primary central nervous system lymphomas (PCNSLs). Therapeutic outcomes in real life-experience of the French Network. Bone Marrow Transplant 57 (6): 966-974, 2022. [PUBMED Abstract]
Jahnke K, Thiel E, Martus P, et al.: Relapse of primary central nervous system lymphoma: clinical features, outcome and prognostic factors. J Neurooncol 80 (2): 159-65, 2006. [PUBMED Abstract]
Han CH, Batchelor TT: Diagnosis and management of primary central nervous system lymphoma. Cancer 123 (22): 4314-4324, 2017. [PUBMED Abstract]
Ghesquieres H, Chevrier M, Laadhari M, et al.: Lenalidomide in combination with intravenous rituximab (REVRI) in relapsed/refractory primary CNS lymphoma or primary intraocular lymphoma: a multicenter prospective ‘proof of concept’ phase II study of the French Oculo-Cerebral lymphoma (LOC) Network and the Lymphoma Study Association (LYSA)†. Ann Oncol 30 (4): 621-628, 2019. [PUBMED Abstract]
Houillier C, Chabrot CM, Moles-Moreau MP, et al.: Rituximab-Lenalidomide-Ibrutinib Combination for Relapsed/Refractory Primary CNS Lymphoma: A Case Series of the LOC Network. Neurology 97 (13): 628-631, 2021. [PUBMED Abstract]
Schmitt AM, Herbrand AK, Fox CP, et al.: Rituximab in primary central nervous system lymphoma-A systematic review and meta-analysis. Hematol Oncol 37 (5): 548-557, 2019. [PUBMED Abstract]
Lionakis MS, Dunleavy K, Roschewski M, et al.: Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary CNS Lymphoma. Cancer Cell 31 (6): 833-843.e5, 2017. [PUBMED Abstract]
Frigault MJ, Dietrich J, Gallagher K, et al.: Safety and efficacy of tisagenlecleucel in primary CNS lymphoma: a phase 1/2 clinical trial. Blood 139 (15): 2306-2315, 2022. [PUBMED Abstract]
Treatment of Intraocular Lymphoma
Retrospective reviews of selected patients with primary intraocular lymphoma and no evidence of disseminated central nervous system (CNS) disease showed that localized therapy with intraocular methotrexate or ocular radiation therapy or systemic therapy with rituximab were effective in clearing lymphoma cells from the eye. However, most patients had subsequent CNS relapse.[1,2][Level of evidence C3] Anecdotal series reported lower relapse rates when high-dose methotrexate was added, but prospective multicenter trials with even retrospective controls do not exist.[1,2][Level of evidence D]
Relapsing disease in the rest of the CNS is treated with the same options listed for primary CNS lymphoma in the brain. In a phase III randomized study of whole-brain radiation therapy, patients with intraocular disease and concomitant brain involvement had a worse prognosis than those with brain involvement alone (19 patients with both, 391 patients with brain involvement only).[3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Grimm SA, Pulido JS, Jahnke K, et al.: Primary intraocular lymphoma: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Ann Oncol 18 (11): 1851-5, 2007. [PUBMED Abstract]
Korfel A, Thiel E, Martus P, et al.: Randomized phase III study of whole-brain radiotherapy for primary CNS lymphoma. Neurology 84 (12): 1242-8, 2015. [PUBMED Abstract]
Key References for Primary Central Nervous System Lymphoma Treatment
These references have been identified by members of the PDQ Adult Treatment Editorial Board as significant in the field of primary central nervous system (CNS) lymphoma treatment. This list is provided to inform users of important studies that have helped shape the current understanding of and treatment options for primary CNS lymphoma. Listed after each reference are the sections within this summary where the reference is cited.
Ferreri AJ, Illerhaus G: The role of autologous stem cell transplantation in primary central nervous system lymphoma. Blood 127 (13): 1642-9, 2016. [PUBMED Abstract]
Korfel A, Thiel E, Martus P, et al.: Randomized phase III study of whole-brain radiotherapy for primary CNS lymphoma. Neurology 84 (12): 1242-8, 2015. [PUBMED Abstract]
Thiel E, Korfel A, Martus P, et al.: High-dose methotrexate with or without whole brain radiotherapy for primary CNS lymphoma (G-PCNSL-SG-1): a phase 3, randomised, non-inferiority trial. Lancet Oncol 11 (11): 1036-47, 2010. [PUBMED Abstract]
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of primary CNS lymphoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Primary Central Nervous System Lymphoma Treatment is:
Eric J. Seifter, MD (Johns Hopkins University)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Primary Central Nervous System Lymphoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/lymphoma/hp/primary-cns-lymphoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389331]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
AIDS was first described in 1981, and the first definitions included certain opportunistic infections, Kaposi sarcoma, and central nervous system (CNS) lymphomas. In 1984, a multicenter study described the clinical spectrum of non-Hodgkin lymphomas (NHLs) in the populations at risk of AIDS.[1] The incidence of NHL has increased in a course almost parallel to that of the AIDS epidemic and accounts for 2% to 3% of newly diagnosed AIDS cases.[2] Since the introduction of highly active antiretroviral therapy (HAART) in the mid-1990s, the incidence of lymphomas has decreased, and outcomes have improved.[3] Higher CD4-positive T-lymphocyte (CD4) counts in the HAART era have been associated with a shift in histological diagnoses. The shift is away from primary effusion lymphoma and primary CNS lymphoma, which occur with the lowest CD4 counts, and toward histologies that occur at higher CD4 counts, such as Burkitt lymphoma and Hodgkin lymphoma (HL).[4–6] In contrast to less-frequent incidences of all the lymphoproliferative disorders in the HAART era, the incidence rate of anal cancer has not changed.[7]
Histology
Pathologically, AIDS-related lymphomas comprise a narrow spectrum of histological types consisting almost exclusively of B-cell tumors of aggressive type. These include:
Diffuse large B-cell lymphoma (including B-cell immunoblastic lymphoma).
Small noncleaved lymphoma, either Burkitt or Burkitt-like.
The HIV-associated lymphomas can be categorized as:
Aggressive B-cell lymphoma (see above).
Primary CNS lymphoma (PCNSL).
Primary effusion lymphoma.
Plasmablastic multicentric Castleman disease.
HL.
HIV-associated Hodgkin lymphoma
Multiple reviews of HL occurring in patients at risk of AIDS have been done;[8,9] however, HL is still not part of the Centers for Disease Control and Prevention (CDC) definition of AIDS because no clear demonstration of its increased incidence in conjunction with HIV has been shown, as is the case for aggressive NHL. The CDC, in conjunction with the San Francisco Department of Public Health, has reported a cohort study in which HIV-infected men had an excess risk that was attributable to the HIV infection in 19.3 cases of HL per 100,000 person-years and 224.9 cases of NHL per 100,000 person-years. Although this report found an excess incidence of HL in HIV-infected homosexual men, additional epidemiologic studies will be needed before the CDC will reconsider HL as an HIV-associated malignancy.[10]
HIV-associated HL presents in an aggressive fashion, often with extranodal or bone marrow involvement.[8,9,11] A distinctive feature of HIV-associated HL is the lesser frequency of mediastinal adenopathy compared with non–HIV-associated HL. Most patients in these series had either mixed cellularity or lymphocyte-depleted HL, expression of Epstein-Barr virus (EBV)-associated proteins in Reed-Sternberg cells, B symptoms, and a median CD4 lymphocyte count of 300/dL or lower.[12] In a retrospective multicenter review of 62 patients, those receiving HAART with chemotherapy had a 74% 2-year overall survival (OS) rate versus a 30% OS rate for those not receiving HAART (P < .001).[13][Level of evidence C1] Among 201 patients with classical HL and HIV positivity, the 2- to 5-year OS rate of 88% to 90% after treatment with ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine), or similar regimens, and HAART, was not significantly different from the OS rate of HIV-negative patients with newly diagnosed HL in two uncontrolled comparisons.[14,15][Level of evidence C3] These studies confirm that patients with HL who were treated with standard regimens and HAART have outcomes that are similar to those of the uninfected population.[16] Furthermore, immune function recovers over the course of 6 to 9 months after completion of chemotherapy.[15]
Primary effusion lymphoma
Primary effusion lymphoma has been associated with Kaposi sarcoma (KS)-associated herpesvirus (KSHV)/human herpes virus type 8 (HHV8).[17,18] Primary effusion lymphoma presents as a liquid phase spreading along serous membranes in the absence of masses or adenopathy.[17] In addition to HHV8, many cases are also associated with EBV. Extension of lymphoma from the effusion to underlying tissue may occur. A series of 20 patients, including 19 treated with modified infusional etoposide, vincristine, and doxorubicin plus cyclophosphamide and prednisone (modEPOCH), had a 3-year cancer-specific survival rate of 47% and a median OS of 22 months.[19][Level of evidence C3]
Multicentric Castleman disease
The plasmablastic type of multicentric Castleman disease is also associated with a coinfection of KSHV/HHV8 and HIV.[20] Patients typically present with fever, night sweats, weight loss, lymphadenopathy, and hepatosplenomegaly. Patients may progress to primary effusion lymphoma or to plasmablastic or anaplastic large cell lymphoma. Anecdotal responses to the anti-CD20 monoclonal antibody rituximab alone (along with HAART), have been reported.[21–24][Level of evidence C3] For a prospective cohort of 84 patients treated with rituximab for HIV and HHV8 multicentric Castleman disease, the 5-year rate of relapse-free survival was 82% (95% confidence interval [CI], 72%–92%), and all patients responded again to rituximab at relapse.[24][Level of evidence C2]
Incidence and Prevention
An international database of 48,000 HIV-seropositive individuals from the United States, Europe, and Australia found a 42% decline in the incidence of NHLs from 1997 to 1999 compared with the same incidences in 1992 to 1996, both for PCNSL and for systemic lymphoma.[25] The introduction of HAART is the proposed explanation for this decline.[26] The diagnosis of AIDS precedes the onset of NHL in approximately 50% of the patients; however, in the other half of the patients, the diagnosis of AIDS is made at the time of the diagnosis of NHL and HIV positivity.[3] The geographic distribution of these lymphomas is also similar to the geographic spread of AIDS. Unlike KS, which has a predilection for homosexual men and appears to be on the decline in incidence, all risk groups appear to have an excess number of NHLs; these risk groups include intravenous drug users and children of HIV-positive individuals.
Clinical Presentation
In general, the clinical setting and response to treatment of patients with AIDS-related lymphoma is very different from that of the non-HIV patients with lymphoma. The HIV-infected individual with aggressive lymphoma usually presents with advanced-stage disease that is frequently extranodal.[27]
Common extranodal sites include:
Bone marrow.
Liver.
Meninges.
Gastrointestinal tract.
Very unusual sites are also characteristic and include:
Anus.
Heart.
Bile duct.
Gingiva.
Muscles.
The clinical course is more aggressive, and the disease is both more extensive and less responsive to chemotherapy. Immunodeficiency and cytopenias, common in these patients at the time of initial presentation, are exacerbated by the administration of chemotherapy. Treatment of the malignancy increases the risk of opportunistic infections, which further compromise the delivery of adequate treatment.
Prognosis and Survival
Prognoses of patients with AIDS-related lymphoma have been associated with:[28]
Stage (i.e., extent of disease, extranodal involvement, lactate dehydrogenase level, and bone marrow involvement).
Age.
Severity of the underlying immunodeficiency (measured by CD4 lymphocyte count in peripheral blood).
Performance status.
Prior AIDS diagnosis (i.e., history of opportunistic infection or KS).
Patients with AIDS-related PCNSL appear to have more severe underlying HIV-related disease than do patients with systemic lymphoma. In one report, this severity was evidenced by patients with PCNSL who had a higher incidence of previously diagnosed AIDS (73% vs. 37%), lower median number of CD4 lymphocytes (30/dL vs. 189/dL), and a worse median survival time (2.5 months vs. 6.0 months).[29] This report also showed that patients with poor risk factors—defined as Karnofsky Performance Status score lower than 70%, history of previously diagnosed AIDS, and bone marrow involvement—had a median survival time of 4.0 months compared with patients in a good prognosis group who had none of these risk factors, and who had a median survival time of 11.3 months.
In another report (NIAID-ACTG-142), prognostic factors were evaluated in a group of 192 patients with newly diagnosed AIDS-related lymphoma who were randomly assigned to receive either low-dose methotrexate, bleomycin, doxorubicin, cyclophosphamide, vincristine, and dexamethasone (m-BACOD) or standard-dose m-BACOD with granulocyte-macrophage colony-stimulating factor.[30] No differences existed between these two treatments in terms of efficacy for disease-free survival, median survival, or risk ratio for death.[30][Level of evidence A1] On multivariate analysis, factors associated with decreased survival included age older than 35 years, history of intravenous drug use, stage III or stage IV disease, and CD4 counts lower than 100 cells/mm3. The International Prognostic Index may also be predictive for survival.[31–33] In a multicenter cohort study of 203 patients, in a multivariable Cox model, response to HAART was independently associated with prolonged survival (relative hazard, 0.32; 95% CI, 0.16–0.62).[34][Level of evidence C2]
References
Ziegler JL, Beckstead JA, Volberding PA, et al.: Non-Hodgkin’s lymphoma in 90 homosexual men. Relation to generalized lymphadenopathy and the acquired immunodeficiency syndrome. N Engl J Med 311 (9): 565-70, 1984. [PUBMED Abstract]
Rabkin CS, Yellin F: Cancer incidence in a population with a high prevalence of infection with human immunodeficiency virus type 1. J Natl Cancer Inst 86 (22): 1711-6, 1994. [PUBMED Abstract]
Little RF, Wilson WH: Update on the Pathogenesis, Diagnosis, and Therapy of AIDS-related Lymphoma. Curr Infect Dis Rep 5 (2): 176-184, 2003. [PUBMED Abstract]
Carbone A, Gloghini A: AIDS-related lymphomas: from pathogenesis to pathology. Br J Haematol 130 (5): 662-70, 2005. [PUBMED Abstract]
Gopal S, Patel MR, Yanik EL, et al.: Temporal trends in presentation and survival for HIV-associated lymphoma in the antiretroviral therapy era. J Natl Cancer Inst 105 (16): 1221-9, 2013. [PUBMED Abstract]
Piketty C, Selinger-Leneman H, Bouvier AM, et al.: Incidence of HIV-related anal cancer remains increased despite long-term combined antiretroviral treatment: results from the french hospital database on HIV. J Clin Oncol 30 (35): 4360-6, 2012. [PUBMED Abstract]
Spina M, Vaccher E, Nasti G, et al.: Human immunodeficiency virus-associated Hodgkin’s disease. Semin Oncol 27 (4): 480-8, 2000. [PUBMED Abstract]
Thompson LD, Fisher SI, Chu WS, et al.: HIV-associated Hodgkin lymphoma: a clinicopathologic and immunophenotypic study of 45 cases. Am J Clin Pathol 121 (5): 727-38, 2004. [PUBMED Abstract]
Hessol NA, Katz MH, Liu JY, et al.: Increased incidence of Hodgkin disease in homosexual men with HIV infection. Ann Intern Med 117 (4): 309-11, 1992. [PUBMED Abstract]
Re A, Casari S, Cattaneo C, et al.: Hodgkin disease developing in patients infected by human immunodeficiency virus results in clinical features and a prognosis similar to those in patients with human immunodeficiency virus-related non-Hodgkin lymphoma. Cancer 92 (11): 2739-45, 2001. [PUBMED Abstract]
Dolcetti R, Boiocchi M, Gloghini A, et al.: Pathogenetic and histogenetic features of HIV-associated Hodgkin’s disease. Eur J Cancer 37 (10): 1276-87, 2001. [PUBMED Abstract]
Hentrich M, Maretta L, Chow KU, et al.: Highly active antiretroviral therapy (HAART) improves survival in HIV-associated Hodgkin’s disease: results of a multicenter study. Ann Oncol 17 (6): 914-9, 2006. [PUBMED Abstract]
Montoto S, Shaw K, Okosun J, et al.: HIV status does not influence outcome in patients with classical Hodgkin lymphoma treated with chemotherapy using doxorubicin, bleomycin, vinblastine, and dacarbazine in the highly active antiretroviral therapy era. J Clin Oncol 30 (33): 4111-6, 2012. [PUBMED Abstract]
Hentrich M, Berger M, Wyen C, et al.: Stage-adapted treatment of HIV-associated Hodgkin lymphoma: results of a prospective multicenter study. J Clin Oncol 30 (33): 4117-23, 2012. [PUBMED Abstract]
Kaplan LD: Management of HIV-associated Hodgkin lymphoma: how far we have come. J Clin Oncol 30 (33): 4056-8, 2012. [PUBMED Abstract]
Simonelli C, Spina M, Cinelli R, et al.: Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: a single-institution study. J Clin Oncol 21 (21): 3948-54, 2003. [PUBMED Abstract]
Nador RG, Cesarman E, Chadburn A, et al.: Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 88 (2): 645-56, 1996. [PUBMED Abstract]
Lurain K, Polizzotto MN, Aleman K, et al.: Viral, immunologic, and clinical features of primary effusion lymphoma. Blood 133 (16): 1753-1761, 2019. [PUBMED Abstract]
Bower M, Newsom-Davis T, Naresh K, et al.: Clinical Features and Outcome in HIV-Associated Multicentric Castleman’s Disease. J Clin Oncol 29 (18): 2481-6, 2011. [PUBMED Abstract]
Goedert JJ: Multicentric Castleman disease: viral and cellular targets for intervention. Blood 102 (8): 2710-11, 2003.
Uldrick TS, Polizzotto MN, Aleman K, et al.: Rituximab plus liposomal doxorubicin in HIV-infected patients with KSHV-associated multicentric Castleman disease. Blood 124 (24): 3544-52, 2014. [PUBMED Abstract]
Pria AD, Pinato D, Roe J, et al.: Relapse of HHV8-positive multicentric Castleman disease following rituximab-based therapy in HIV-positive patients. Blood 129 (15): 2143-2147, 2017. [PUBMED Abstract]
International Collaboration on HIV and Cancer: Highly active antiretroviral therapy and incidence of cancer in human immunodeficiency virus-infected adults. J Natl Cancer Inst 92 (22): 1823-30, 2000. [PUBMED Abstract]
Stebbing J, Gazzard B, Mandalia S, et al.: Antiretroviral treatment regimens and immune parameters in the prevention of systemic AIDS-related non-Hodgkin’s lymphoma. J Clin Oncol 22 (11): 2177-83, 2004. [PUBMED Abstract]
Sparano JA: Clinical aspects and management of AIDS-related lymphoma. Eur J Cancer 37 (10): 1296-305, 2001. [PUBMED Abstract]
Bower M, Gazzard B, Mandalia S, et al.: A prognostic index for systemic AIDS-related non-Hodgkin lymphoma treated in the era of highly active antiretroviral therapy. Ann Intern Med 143 (4): 265-73, 2005. [PUBMED Abstract]
Levine AM, Sullivan-Halley J, Pike MC, et al.: Human immunodeficiency virus-related lymphoma. Prognostic factors predictive of survival. Cancer 68 (11): 2466-72, 1991. [PUBMED Abstract]
Kaplan LD, Straus DJ, Testa MA, et al.: Low-dose compared with standard-dose m-BACOD chemotherapy for non-Hodgkin’s lymphoma associated with human immunodeficiency virus infection. National Institute of Allergy and Infectious Diseases AIDS Clinical Trials Group. N Engl J Med 336 (23): 1641-8, 1997. [PUBMED Abstract]
Navarro JT, Ribera JM, Oriol A, et al.: International prognostic index is the best prognostic factor for survival in patients with AIDS-related non-Hodgkin’s lymphoma treated with CHOP. A multivariate study of 46 patients. Haematologica 83 (6): 508-13, 1998. [PUBMED Abstract]
Rossi G, Donisi A, Casari S, et al.: The International Prognostic Index can be used as a guide to treatment decisions regarding patients with human immunodeficiency virus-related systemic non-Hodgkin lymphoma. Cancer 86 (11): 2391-7, 1999. [PUBMED Abstract]
Straus DJ, Huang J, Testa MA, et al.: Prognostic factors in the treatment of human immunodeficiency virus-associated non-Hodgkin’s lymphoma: analysis of AIDS Clinical Trials Group protocol 142–low-dose versus standard-dose m-BACOD plus granulocyte-macrophage colony-stimulating factor. National Institute of Allergy and Infectious Diseases. J Clin Oncol 16 (11): 3601-6, 1998. [PUBMED Abstract]
Hoffmann C, Wolf E, Fätkenheuer G, et al.: Response to highly active antiretroviral therapy strongly predicts outcome in patients with AIDS-related lymphoma. AIDS 17 (10): 1521-9, 2003. [PUBMED Abstract]
Cellular Classification of AIDS-Related Lymphoma
Pathologically, AIDS-related lymphomas comprise a narrow spectrum of histological types consisting almost exclusively of B-cell tumors of aggressive type. These include:
Diffuse large B-cell lymphoma (B-cell immunoblastic lymphoma).
Small noncleaved lymphoma, either Burkitt or Burkitt-like.
AIDS-related lymphomas, though usually of B-cell origin as demonstrated by immunoglobulin heavy-chain gene rearrangement studies, have also been shown to be oligoclonal, polyclonal, and monoclonal in origin. Although HIV does not appear to have a direct etiologic role, HIV infection does lead to an altered immunologic milieu. HIV generally infects T lymphocytes with the loss of regulation function that leads to hypergammaglobulinemia and polyclonal B-cell hyperplasia. B cells are not the targets of HIV infection. Instead, Epstein-Barr virus (EBV) is thought to be at least a cofactor in the etiology of some of these lymphomas. The EBV genome has been detected in most patients with AIDS-related lymphomas; molecular analysis suggests that the cells were infected before clonal proliferation began.[1] The rare primary effusion lymphoma consistently harbors human herpes virus type 8 and frequently contains EBV.[2] HIV-related T-cell lymphomas have also been identified and appear to be associated with EBV infection.[3]
References
Thorley-Lawson DA, Gross A: Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med 350 (13): 1328-37, 2004. [PUBMED Abstract]
Simonelli C, Spina M, Cinelli R, et al.: Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: a single-institution study. J Clin Oncol 21 (21): 3948-54, 2003. [PUBMED Abstract]
Thomas JA, Cotter F, Hanby AM, et al.: Epstein-Barr virus-related oral T-cell lymphoma associated with human immunodeficiency virus immunosuppression. Blood 81 (12): 3350-6, 1993. [PUBMED Abstract]
Stage Information for AIDS-Related Lymphoma
Although stage is important in selecting the treatment of patients with non-Hodgkin lymphoma (NHL) who do not have AIDS, most patients with AIDS-related lymphomas have far-advanced disease.
Staging Subclassification System
Lugano classification
The American Joint Committee on Cancer (AJCC) has adopted the Lugano classification to evaluate and stage lymphoma.[1] The Lugano classification system replaces the Ann Arbor classification system, which was adopted in 1971 at the Ann Arbor Conference,[2] with some modifications 18 years later from the Cotswolds meeting.[3,4]
Table 1. Lugano Classification for Hodgkin and Non-Hodgkin Lymphomaa
Stage
Stage Description
Illustration
CSF = cerebrospinal fluid; CT = computed tomography; DLBCL = diffuse large B-cell lymphoma; NHL = non-Hodgkin lymphoma.
aHodgkin and Non-Hodgkin Lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 937–58.
bStage II bulky may be considered either early or advanced stage based on lymphoma histology and prognostic factors.
cThe definition of disease bulk varies according to lymphoma histology. In the Lugano classification, bulk ln Hodgkin lymphoma is defined as a mass greater than one-third of the thoracic diameter on CT of the chest or a mass >10 cm. For NHL, the recommended definitions of bulk vary by lymphoma histology. In follicular lymphoma, 6 cm has been suggested based on the Follicular Lymphoma International Prognostic Index-2 and its validation. In DLBCL, cutoffs ranging from 5 cm to 10 cm have been used, although 10 cm is recommended.
Limited stage
I
Involvement of a single lymphatic site (i.e., nodal region, Waldeyer’s ring, thymus, or spleen).
Diffuse or disseminated involvement of one or more extralymphatic organs, with or without associated lymph node involvement; or noncontiguous extralymphatic organ involvement in conjunction with nodal stage II disease; or any extralymphatic organ involvement in nodal stage III disease. Stage IV includes any involvement of the CSF, bone marrow, liver, or lungs (other than by direct extension in stage IIE disease).
Note: Hodgkin lymphoma uses A or B designation with stage group. A/B is no longer used in NHL.
Occasionally, specialized staging systems are used. The physician should be aware of the system used in a specific report.
The E designation is used when extranodal lymphoid malignancies arise in tissues separate from, but near, the major lymphatic aggregates. Stage IV refers to disease that is diffusely spread throughout an extranodal site, such as the liver. If pathological proof of involvement of one or more extralymphatic sites has been documented, the symbol for the site of involvement, followed by a plus sign (+), is listed.
Table 2. Notation for Identification of Sites
N = nodes
H = liver
L = lung
M = marrow
S = spleen
P = pleura
O = bone
D = skin
Current practice assigns a clinical stage (CS) based on the findings of the clinical evaluation and a pathological stage (PS) based on the findings made as a result of invasive procedures beyond the initial biopsy.
For example, on percutaneous biopsy, a patient with inguinal adenopathy and a positive lymphangiogram without systemic symptoms might be found to have involvement of the liver and bone marrow. The precise stage of such a patient would be CS IIA, PS IVA(H+)(M+).
A number of other factors that are not included in the above staging system are important for the staging and prognosis of patients with NHL. These factors include:
Age.
Performance status (PS).
Tumor size.
Lactate dehydrogenase (LDH) values.
The number of extranodal sites.
References
Hodgkin and non-Hodgkin lymphoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 937–58.
Carbone PP, Kaplan HS, Musshoff K, et al.: Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res 31 (11): 1860-1, 1971. [PUBMED Abstract]
Lister TA, Crowther D, Sutcliffe SB, et al.: Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol 7 (11): 1630-6, 1989. [PUBMED Abstract]
National Cancer Institute sponsored study of classifications of non-Hodgkin’s lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin’s Lymphoma Pathologic Classification Project. Cancer 49 (10): 2112-35, 1982. [PUBMED Abstract]
Treatment Option Overview for AIDS-Related Lymphoma
The treatment of patients with AIDS-related lymphomas presents the challenge of integrating therapy appropriate for the stage and histological subset of malignant lymphoma with the limitations imposed by HIV infection.[1] In addition to antitumor therapy, essential components of an optimal non-Hodgkin lymphoma treatment strategy include:[2,3]
Highly active antiretroviral therapy.
Prophylaxis for opportunistic infections.
Rapid recognition and treatment of intercurrent infections.
Patients with HIV positivity and underlying immunodeficiency have poor bone marrow reserve, which compromises the potential for drug dose intensity. Intercurrent opportunistic infection is a risk that may also lead to a decrease in drug delivery. Furthermore, chemotherapy itself compromises the immune system and increases the likelihood of opportunistic infection.
Treatment of AIDS-Related Peripheral/Systemic Lymphoma
The treatment of AIDS-related lymphomas involves overcoming several problems. These are all aggressive lymphomas, which by definition are diffuse large cell/immunoblastic lymphoma or small noncleaved cell lymphoma (Burkitt lymphoma). These lymphomas frequently involve the bone marrow and central nervous system (CNS) and, therefore, are usually in an advanced stage. In addition, the immunodeficiency of AIDS and the leukopenia that is commonly seen with HIV infection makes the use of immunosuppressive chemotherapy challenging.
The introduction of highly active antiretroviral therapy (HAART) has led to a marked reduction in opportunistic infections, prolonged survival with HIV infection, and a median overall survival (OS) for patients with AIDS-related lymphoma that is comparable with the outcome in the nonimmunosuppressed population.[1–7][Level of evidence C3] The use of HAART has also allowed standard-dose and even intensive chemotherapy regimens to be given with reasonable safety to patients with AIDS-related lymphomas, which is comparable with the outcome in patients without HIV.[1–10]
Several prospective nonrandomized trials and pooled individual data from 19 prospective trials that included 1,546 patients show that the addition of rituximab to combination chemotherapy improves the complete response rate, progression-free survival, and OS.[3–6][Level of evidence C3] Several other prospective nonrandomized trials and the pooled individual data from the same 1,546 patients also show that infusional EPOCH (infusional etoposide, infusional vincristine, infusional doxorubicin, cyclophosphamide, and prednisone) produced better outcomes than did CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) (hazard ratioOS, 0.33; P = .03).[3,6,7][Level of evidence C3] Concurrent use of HAART with the infusional EPOCH regimen is controversial; one group advocated for HAART after completion of chemotherapy,[7] while others allowed concurrent therapy.[6]
For patients with Burkitt lymphoma, dose-modified regimens such as R-CODOX (cyclophosphamide, doxorubicin, vincristine, methotrexate, cytarabine and rituximab)-M/IVAC (ifosfamide, etoposide, and high-dose cytarabine) or R-EPOCH (etoposide, prednisone, vincristine, and doxorubicin in combination with rituximab) have shown good results with concurrent or sequential HAART.[7,11,12]
Patients at risk of subsequent CNS involvement include those with bone marrow involvement or those with EBV identified in the primary tumor or in the cerebrospinal fluid (i.e., by polymerase chain reaction).[13,14] Intrathecal chemotherapy is usually considered for patients who are at higher risk of CNS involvement.
Highly selected patients with resistant or relapsed lymphoma after first-line chemotherapy and with continued responsiveness to HAART underwent second-line chemotherapy followed by high-dose therapy and autologous peripheral stem cell transplant. Long-term survivors have been reported anecdotally for these highly selected patients who relapsed.[15–18][Level of evidence C3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Mounier N, Spina M, Gabarre J, et al.: AIDS-related non-Hodgkin lymphoma: final analysis of 485 patients treated with risk-adapted intensive chemotherapy. Blood 107 (10): 3832-40, 2006. [PUBMED Abstract]
Weiss R, Mitrou P, Arasteh K, et al.: Acquired immunodeficiency syndrome-related lymphoma: simultaneous treatment with combined cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy and highly active antiretroviral therapy is safe and improves survival–results of the German Multicenter Trial. Cancer 106 (7): 1560-8, 2006. [PUBMED Abstract]
Barta SK, Xue X, Wang D, et al.: Treatment factors affecting outcomes in HIV-associated non-Hodgkin lymphomas: a pooled analysis of 1546 patients. Blood 122 (19): 3251-62, 2013. [PUBMED Abstract]
Wyen C, Jensen B, Hentrich M, et al.: Treatment of AIDS-related lymphomas: rituximab is beneficial even in severely immunosuppressed patients. AIDS 26 (4): 457-64, 2012. [PUBMED Abstract]
Levine AM, Noy A, Lee JY, et al.: Pegylated liposomal doxorubicin, rituximab, cyclophosphamide, vincristine, and prednisone in AIDS-related lymphoma: AIDS Malignancy Consortium Study 047. J Clin Oncol 31 (1): 58-64, 2013. [PUBMED Abstract]
Sparano JA, Lee JY, Kaplan LD, et al.: Rituximab plus concurrent infusional EPOCH chemotherapy is highly effective in HIV-associated B-cell non-Hodgkin lymphoma. Blood 115 (15): 3008-16, 2010. [PUBMED Abstract]
Dunleavy K, Little RF, Pittaluga S, et al.: The role of tumor histogenesis, FDG-PET, and short-course EPOCH with dose-dense rituximab (SC-EPOCH-RR) in HIV-associated diffuse large B-cell lymphoma. Blood 115 (15): 3017-24, 2010. [PUBMED Abstract]
Ratner L, Lee J, Tang S, et al.: Chemotherapy for human immunodeficiency virus-associated non-Hodgkin’s lymphoma in combination with highly active antiretroviral therapy. J Clin Oncol 19 (8): 2171-8, 2001. [PUBMED Abstract]
Wang ES, Straus DJ, Teruya-Feldstein J, et al.: Intensive chemotherapy with cyclophosphamide, doxorubicin, high-dose methotrexate/ifosfamide, etoposide, and high-dose cytarabine (CODOX-M/IVAC) for human immunodeficiency virus-associated Burkitt lymphoma. Cancer 98 (6): 1196-205, 2003. [PUBMED Abstract]
Cortes J, Thomas D, Rios A, et al.: Hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone and highly active antiretroviral therapy for patients with acquired immunodeficiency syndrome-related Burkitt lymphoma/leukemia. Cancer 94 (5): 1492-9, 2002. [PUBMED Abstract]
Noy A, Kaplan L, Lee J: Feasibility and toxicity of a modified dose intensive R-CODOX-M/IVAC for HIV-associated Burkitt and atypical Burkitt lymphoma (BL): preliminary results of a prospective multicenter phase II trial of the AIDS Malignancy Consortium (AMC). [Abstract] Blood 114 (22): A-3673, 2009.
Cingolani A, Gastaldi R, Fassone L, et al.: Epstein-Barr virus infection is predictive of CNS involvement in systemic AIDS-related non-Hodgkin’s lymphomas. J Clin Oncol 18 (19): 3325-30, 2000. [PUBMED Abstract]
Scadden DT: Epstein-Barr virus, the CNS, and AIDS-related lymphomas: as close as flame to smoke. J Clin Oncol 18 (19): 3323-4, 2000. [PUBMED Abstract]
Re A, Michieli M, Casari S, et al.: High-dose therapy and autologous peripheral blood stem cell transplantation as salvage treatment for AIDS-related lymphoma: long-term results of the Italian Cooperative Group on AIDS and Tumors (GICAT) study with analysis of prognostic factors. Blood 114 (7): 1306-13, 2009. [PUBMED Abstract]
Krishnan A, Molina A, Zaia J, et al.: Durable remissions with autologous stem cell transplantation for high-risk HIV-associated lymphomas. Blood 105 (2): 874-8, 2005. [PUBMED Abstract]
Costello RT, Zerazhi H, Charbonnier A, et al.: Intensive sequential chemotherapy with hematopoietic growth factor support for non-Hodgkin lymphoma in patients infected with the human immunodeficiency virus. Cancer 100 (4): 667-76, 2004. [PUBMED Abstract]
Balsalobre P, Díez-Martín JL, Re A, et al.: Autologous stem-cell transplantation in patients with HIV-related lymphoma. J Clin Oncol 27 (13): 2192-8, 2009. [PUBMED Abstract]
Treatment of AIDS-Related Primary Central Nervous System Lymphoma
As with other AIDS-related lymphomas, primary central nervous system (CNS) lymphoma (PCNSL) is an aggressive B-cell neoplasm, either diffuse large B-cell or diffuse immunoblastic non-Hodgkin lymphoma (a subtype of diffuse large B-cell lymphoma). AIDS-related PCNSL has been reported to have a 100% association with Epstein-Barr virus (EBV).[1] These patients usually have evidence of low CD4-positive T lymphocyte counts, high HIV viral load, severe debilitation, and focal neurological symptoms such as seizures, changes in mental status, and paralysis.
Computed tomographic scans show contrast-enhancing mass lesions that may not always be distinguished from other CNS diseases, such as toxoplasmosis, that occur in patients with AIDS.[2] Magnetic resonance imaging studies using gadolinium contrast may be a more useful initial diagnostic tool in differentiating lymphoma from cerebral toxoplasmosis or progressive multifocal leukoencephalopathy. Lymphoma tends to present with large lesions, which are enhanced by gadolinium. In cerebral toxoplasmosis, ring enhancement is very common, lesions tend to be smaller, and multiple lesions are seen.[3–5] Use of positron emission scanning has demonstrated an improved ability to distinguish PCNSL from toxoplasmosis.[6,7] PCNSL has an increased uptake while toxoplasmosis lesions are metabolically inactive. Antibodies against toxoplasmosis may also be very useful because most cerebral toxoplasmosis occurs as a consequence of reactivity of a previous infection. If the immunoglobulin G titer is less than 1:4, the disease is unlikely to be toxoplasmotic. A lumbar puncture may be useful to detect as many as 23% of patients with malignant cells in their cerebrospinal fluid (CSF). Evaluating the CSF for EBV DNA may be a useful lymphoma-specific tool because EBV is present in all patients with PCNSL. Despite the many evaluations, however, most patients with PCNSL require a pathological diagnosis.[8–10] Diagnosis is made by biopsy. Sometimes, a biopsy is attempted only after failure of antibiotics for toxoplasmosis, which will produce clinical and radiographic improvement within 1 to 3 weeks in patients with cerebral toxoplasmosis.[11]
Radiation therapy alone has usually been used in this group of patients. With doses in the range of 35 Gy to 40 Gy, median duration of survival has been only 72 to 119 days.[2,12,13] Survival is longer in younger patients with better performance status and the absence of opportunistic infection.[14] In the highly active antiretroviral therapy (HAART) era, a median survival of 18 months has been seen with radiation therapy alone.[15] An anecdotal report using HAART and high-dose methotrexate for patients with AIDS-related PCNSL showed a median survival that had not been reached with a median follow-up of 27 months.[16] Most patients respond to treatment by showing partial improvement in neurological symptoms. Autopsies have revealed that these patients die of opportunistic infections as well as tumor progression. Treatment of these patients is also complicated by other AIDS-related CNS infections, including subacute AIDS encephalitis, cytomegalovirus encephalitis, and toxoplasmosis encephalitis. Spontaneous remissions have been reported after HAART.[17]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
MacMahon EM, Glass JD, Hayward SD, et al.: Epstein-Barr virus in AIDS-related primary central nervous system lymphoma. Lancet 338 (8773): 969-73, 1991. [PUBMED Abstract]
Goldstein JD, Dickson DW, Moser FG, et al.: Primary central nervous system lymphoma in acquired immune deficiency syndrome. A clinical and pathologic study with results of treatment with radiation. Cancer 67 (11): 2756-65, 1991. [PUBMED Abstract]
Nyberg DA, Federle MP: AIDS-related Kaposi sarcoma and lymphomas. Semin Roentgenol 22 (1): 54-65, 1987. [PUBMED Abstract]
Fine HA, Mayer RJ: Primary central nervous system lymphoma. Ann Intern Med 119 (11): 1093-104, 1993. [PUBMED Abstract]
Ciricillo SF, Rosenblum ML: Use of CT and MR imaging to distinguish intracranial lesions and to define the need for biopsy in AIDS patients. J Neurosurg 73 (5): 720-4, 1990. [PUBMED Abstract]
Hoffman JM, Waskin HA, Schifter T, et al.: FDG-PET in differentiating lymphoma from nonmalignant central nervous system lesions in patients with AIDS. J Nucl Med 34 (4): 567-75, 1993. [PUBMED Abstract]
Pierce MA, Johnson MD, Maciunas RJ, et al.: Evaluating contrast-enhancing brain lesions in patients with AIDS by using positron emission tomography. Ann Intern Med 123 (8): 594-8, 1995. [PUBMED Abstract]
Cinque P, Brytting M, Vago L, et al.: Epstein-Barr virus DNA in cerebrospinal fluid from patients with AIDS-related primary lymphoma of the central nervous system. Lancet 342 (8868): 398-401, 1993. [PUBMED Abstract]
Cingolani A, De Luca A, Larocca LM, et al.: Minimally invasive diagnosis of acquired immunodeficiency syndrome-related primary central nervous system lymphoma. J Natl Cancer Inst 90 (5): 364-9, 1998. [PUBMED Abstract]
Yarchoan R, Jaffe ES, Little R: Diagnosing central nervous system lymphoma in the setting of AIDS: a step forward. J Natl Cancer Inst 90 (5): 346-7, 1998. [PUBMED Abstract]
Mathews C, Barba D, Fullerton SC: Early biopsy versus empiric treatment with delayed biopsy of non-responders in suspected HIV-associated cerebral toxoplasmosis: a decision analysis. AIDS 9 (11): 1243-50, 1995. [PUBMED Abstract]
Baumgartner JE, Rachlin JR, Beckstead JH, et al.: Primary central nervous system lymphomas: natural history and response to radiation therapy in 55 patients with acquired immunodeficiency syndrome. J Neurosurg 73 (2): 206-11, 1990. [PUBMED Abstract]
Remick SC, Diamond C, Migliozzi JA, et al.: Primary central nervous system lymphoma in patients with and without the acquired immune deficiency syndrome. A retrospective analysis and review of the literature. Medicine (Baltimore) 69 (6): 345-60, 1990. [PUBMED Abstract]
Corn BW, Donahue BR, Rosenstock JG, et al.: Performance status and age as independent predictors of survival among AIDS patients with primary CNS lymphoma: a multivariate analysis of a multi-institutional experience. Cancer J Sci Am 3 (1): 52-6, 1997 Jan-Feb. [PUBMED Abstract]
Hoffmann C, Tabrizian S, Wolf E, et al.: Survival of AIDS patients with primary central nervous system lymphoma is dramatically improved by HAART-induced immune recovery. AIDS 15 (16): 2119-27, 2001. [PUBMED Abstract]
Gupta NK, Nolan A, Omuro A, et al.: Long-term survival in AIDS-related primary central nervous system lymphoma. Neuro Oncol 19 (1): 99-108, 2017. [PUBMED Abstract]
McGowan JP, Shah S: Long-term remission of AIDS-related primary central nervous system lymphoma associated with highly active antiretroviral therapy. AIDS 12 (8): 952-4, 1998. [PUBMED Abstract]
Latest Updates to This Summary (09/18/2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of AIDS-related lymphoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for AIDS-Related Lymphoma Treatment are:
Eric J. Seifter, MD (Johns Hopkins University)
Minh Tam Truong, MD (Boston University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ AIDS-Related Lymphoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/lymphoma/hp/aids-related-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389186]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Non-Hodgkin Lymphoma Treatment During Pregnancy (PDQ®)–Health Professional Version
General Information About Non-Hodgkin Lymphoma Treatment During Pregnancy
Non-Hodgkin lymphoma (NHL) is more common in older patients, with a median age at diagnosis of 68 years.[1] Hodgkin lymphoma is most common in early adulthood and in patients older than 65 years.[2] This age distribution may explain why there are fewer reports of NHL in pregnant patients.[3]
References
Surveillance, Epidemiology, and End Results Program: SEER Cancer Stat Facts: Non-Hodgkin Lymphoma. Bethesda, Md: National Cancer Institute, DCCPS, Surveillance Research Program. Available online. Last accessed December 16, 2024.
Surveillance, Epidemiology, and End Results Program: SEER Cancer Stat Facts: Hodgkin Lymphoma. Bethesda, Md: National Cancer Institute. Available online. Last accessed December 16, 2024.
Stage Information for Non-Hodgkin Lymphoma During Pregnancy
To avoid exposure to ionizing radiation, magnetic resonance imaging is the preferred tool for staging evaluation.[1] For more information, see the Stage Information for Indolent B-Cell Non-Hodgkin Lymphoma section in Indolent B-Cell Non-Hodgkin Lymphoma Treatment.
References
Nicklas AH, Baker ME: Imaging strategies in the pregnant cancer patient. Semin Oncol 27 (6): 623-32, 2000. [PUBMED Abstract]
Treatment Option Overview for Non-Hodgkin Lymphoma During Pregnancy
Table 1. Treatment Options for Non-Hodgkin Lymphoma (NHL) During Pregnancy
Treatment of Indolent Non-Hodgkin Lymphoma During Pregnancy
Treatment may be delayed for women with an indolent non-Hodgkin lymphoma.
Treatment of Aggressive Non-Hodgkin Lymphoma During Pregnancy
Immediate Therapy
According to anecdotal case series, most non-Hodgkin lymphomas (NHLs) in pregnant patients are aggressive, and delay of therapy until after delivery appears to have poor outcomes.[1–4] Consequently, some investigators favor immediate therapy, even during pregnancy.[4] In a review of 121 patient case reports from 74 papers, one-half of the patients had very aggressive lymphomas, such as Burkitt lymphoma, and one-half of the patients had involvement of the breast, ovaries, uterus, or placenta.[5] One-half of the patients received therapy antepartum, and the 6-month survival rate was 53%, with a live-birth rate of 83%.[5][Level of evidence C3]
A multicenter retrospective analysis of 50 patients described pregnancy termination in 3 patients, deferral of therapy to postpartum in 15 patients (median 30 weeks gestation), and antenatal therapy in the remaining 32 patients (median 21 weeks gestation, all done after the first trimester).[6] With a median follow-up of 41 months, the 3-year progression-free survival rate was 53%, and the overall survival rate was 82%, using R-CHOP (rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone) or modifications of this regimen.[6][Level of evidence C3]
Early Delivery, When Feasible
For some women, early delivery, when feasible, may minimize or avoid fetal exposure to chemotherapy or radiation therapy.
Termination of Pregnancy
Termination of pregnancy in the first trimester may allow immediate therapy for women with aggressive NHL.
Effects of Treatment on Children
Evidence (treatment effect on children exposed in utero):
With follow-up ranging from several months to 11 years, children who were exposed to high-dose doxorubicin-containing combination chemotherapy in utero (especially during the second and third trimester) had normal growth and no evidence of congenital malformations.[4,7–9] For most of the chemotherapeutic agents used for the treatment of NHL, there are no data regarding long-term effects on children exposed in utero.
In one anecdotal case, a newborn exposed to a rituximab-containing regimen in utero was born with no circulating B lymphocytes. The newborn was otherwise healthy and recovered the circulating B lymphocytes by age 6 months with no unusual or persisting intercurrent infections.[10]
Steiner-Salz D, Yahalom J, Samuelov A, et al.: Non-Hodgkin’s lymphoma associated with pregnancy. A report of six cases, with a review of the literature. Cancer 56 (8): 2087-91, 1985. [PUBMED Abstract]
Spitzer M, Citron M, Ilardi CF, et al.: Non-Hodgkin’s lymphoma during pregnancy. Gynecol Oncol 43 (3): 309-12, 1991. [PUBMED Abstract]
Gelb AB, van de Rijn M, Warnke RA, et al.: Pregnancy-associated lymphomas. A clinicopathologic study. Cancer 78 (2): 304-10, 1996. [PUBMED Abstract]
Horowitz NA, Benyamini N, Wohlfart K, et al.: Reproductive organ involvement in non-Hodgkin lymphoma during pregnancy: a systematic review. Lancet Oncol 14 (7): e275-82, 2013. [PUBMED Abstract]
Evens AM, Advani R, Press OW, et al.: Lymphoma occurring during pregnancy: antenatal therapy, complications, and maternal survival in a multicenter analysis. J Clin Oncol 31 (32): 4132-9, 2013. [PUBMED Abstract]
Avilés A, Díaz-Maqueo JC, Torras V, et al.: Non-Hodgkin’s lymphomas and pregnancy: presentation of 16 cases. Gynecol Oncol 37 (3): 335-7, 1990. [PUBMED Abstract]
Moore DT, Taslimi MM: Multi-agent chemotherapy in a case of non-Hodgkin’s lymphoma in second trimester of pregnancy. J Tenn Med Assoc 84 (9): 435-6, 1991. [PUBMED Abstract]
Nantel S, Parboosingh J, Poon MC: Treatment of an aggressive non-Hodgkin’s lymphoma during pregnancy with MACOP-B chemotherapy. Med Pediatr Oncol 18 (2): 143-5, 1990. [PUBMED Abstract]
Mandal PK, Dolai TK, Bagchi B, et al.: B cell suppression in newborn following treatment of pregnant diffuse large B-cell lymphoma patient with rituximab containing regimen. Indian J Pediatr 81 (10): 1092-4, 2014. [PUBMED Abstract]
Latest Updates to This Summary (02/18/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of non-Hodgkin lymphoma during pregnancy. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Non-Hodgkin Lymphoma Treatment During Pregnancy are:
Eric J. Seifter, MD (Johns Hopkins University)
Cole H. Sterling, MD (Johns Hopkins University)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Hodgkin Lymphoma Treatment During Pregnancy (PDQ®)–Health Professional Version
General Information About Hodgkin Lymphoma Treatment During Pregnancy
Hodgkin lymphoma (HL) primarily affects young women, some of whom may be pregnant. When treating a pregnant woman, an oncologist will provide therapy that minimizes risk to the fetus. Treatment choice must be individualized, taking the following into consideration:
The mother’s wishes.
The severity and aggressiveness of the HL.
The trimester of the pregnancy.
Stage Information for Hodgkin Lymphoma During Pregnancy
To avoid exposing a pregnant woman to ionizing radiation, magnetic resonance imaging is the preferred method for staging evaluation.[1] The presenting stage, clinical behavior, prognosis, and histological subtypes of Hodgkin lymphoma (HL) in pregnant women do not differ from those in nonpregnant women during their childbearing years.[2] For more information, see the Stage Information for HL section in Hodgkin Lymphoma Treatment.
References
Nicklas AH, Baker ME: Imaging strategies in the pregnant cancer patient. Semin Oncol 27 (6): 623-32, 2000. [PUBMED Abstract]
Gelb AB, van de Rijn M, Warnke RA, et al.: Pregnancy-associated lymphomas. A clinicopathologic study. Cancer 78 (2): 304-10, 1996. [PUBMED Abstract]
Treatment Options for Hodgkin Lymphoma During Pregnancy
Treatment options for Hodgkin lymphoma (HL) during pregnancy include:
Watchful waiting.
Radiation therapy.
Chemotherapy.
In one study, the 20-year survival rate of pregnant women with HL did not differ from the 20-year survival rate of nonpregnant women who were matched for similar stage of disease, age at diagnosis, and calendar year of treatment.[1]
The long-term effects on children after chemotherapy exposure in utero are unknown.[1–5]
Based on anecdotal series, there is no evidence that a pregnancy after completion of therapy increases the relapse rate for patients in remission.[6,7]
Therapy During the First Trimester
HL that is diagnosed in the first trimester of pregnancy does not constitute an absolute indication for therapeutic abortion. Treatment options for each patient must take into account disease stage, rapidity of growth of the lymphoma, and the patient’s wishes.[8]
Watchful waiting
If the HL presents in early stage above the diaphragm and is growing slowly, patients can be observed carefully, with plans to induce delivery early and proceed with definitive therapy.[9]
Radiation therapy
Alternatively, these patients can receive radiation therapy with proper shielding.[10–13] Investigators at the MD Anderson Cancer Center reported no congenital abnormalities in 16 babies delivered after the mothers had received supradiaphragmatic radiation while the uterus was shielded with five half-value layers of lead.[14] Because of theoretical risks of the fetus developing future malignancies from even minimal scattered radiation doses outside the radiation field, postponing radiation therapy—if possible, until after delivery—should be considered.[15]
Chemotherapy
Evidence (chemotherapy during the first trimester):
Chemotherapy that is administered during the first trimester has been associated with congenital abnormalities in as many as 33% of infants.[2,16] Consequently, some women may opt to continue the pregnancy and agree to radiation therapy or chemotherapy if immediate treatment is required after the first trimester.
A multicenter retrospective analysis of 40 patients described pregnancy termination in 3 patients, deferral of therapy to the postpartum period in 13 patients (median 30-week gestation), and antenatal therapy given to the remaining 24 patients (median 21-week gestation, all done after the first trimester).[17]
With a median follow-up of 41 months, the 3-year progression-free survival (PFS) rate was 85%, and the overall survival (OS) rate was 97%, often with the use of ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine).[17][Level of evidence C3]
A retrospective analysis of 39 patients from the MD Anderson Cancer Center described pregnancy termination in 3 patients, deferral of therapy to the postpartum period in 12 patients, and antenatal therapy given to 24 patients.[18]
Two women had a miscarriage after receiving doxorubicin-based chemotherapy during the first trimester.
With a median follow-up of 68 months from diagnosis, the 5-year PFS rate was 75%, and the OS rate was 82%. These rates did not differ between the antenatal and postpartum timing of therapy.[18][Level of evidence C3]
Therapy Later in Pregnancy
Watchful waiting
In the second half of pregnancy, patients can be observed carefully, and therapy can be postponed until induction of delivery at 32 to 36 weeks.[4,5,16]
Radiation therapy
As an alternative, a short course of radiation therapy can be used before delivery in cases of respiratory compromise caused by a rapidly enlarging mediastinal mass.
Chemotherapy
If chemotherapy is mandatory before delivery—such as for patients with symptomatic advanced-stage disease—vinblastine alone, given intravenously at 6 mg/m² every 2 weeks until induction of delivery, may be considered because it has not been associated with fetal abnormalities in the second half of pregnancy.[4,5] Combination chemotherapy with ABVD appears to be safe in the second half of pregnancy.[3] If chemotherapy is required after the first trimester, many clinicians prefer the combination of drugs over single-agent drugs or radiation therapy. Steroids are used both for their antitumor effect and for hastening fetal pulmonary maturity.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Lishner M, Zemlickis D, Degendorfer P, et al.: Maternal and foetal outcome following Hodgkin’s disease in pregnancy. Br J Cancer 65 (1): 114-7, 1992. [PUBMED Abstract]
Thomas PR, Biochem D, Peckham MJ: The investigation and management of Hodgkin’s disease in the pregnant patient. Cancer 38 (3): 1443-51, 1976. [PUBMED Abstract]
Avilés A, Díaz-Maqueo JC, Talavera A, et al.: Growth and development of children of mothers treated with chemotherapy during pregnancy: current status of 43 children. Am J Hematol 36 (4): 243-8, 1991. [PUBMED Abstract]
Jacobs C, Donaldson SS, Rosenberg SA, et al.: Management of the pregnant patient with Hodgkin’s disease. Ann Intern Med 95 (6): 669-75, 1981. [PUBMED Abstract]
Nisce LZ, Tome MA, He S, et al.: Management of coexisting Hodgkin’s disease and pregnancy. Am J Clin Oncol 9 (2): 146-51, 1986. [PUBMED Abstract]
Weibull CE, Eloranta S, Smedby KE, et al.: Pregnancy and the Risk of Relapse in Patients Diagnosed With Hodgkin Lymphoma. J Clin Oncol 34 (4): 337-44, 2016. [PUBMED Abstract]
Gaudio F, Nardelli C, Masciandaro P, et al.: Pregnancy rate and outcome of pregnancies in long-term survivors of Hodgkin’s lymphoma. Ann Hematol 98 (8): 1947-1952, 2019. [PUBMED Abstract]
Koren G, Weiner L, Lishner M, et al.: Cancer in pregnancy: identification of unanswered questions on maternal and fetal risks. Obstet Gynecol Surv 45 (8): 509-14, 1990. [PUBMED Abstract]
Anselmo AP, Cavalieri E, Enrici RM, et al.: Hodgkin’s disease during pregnancy: diagnostic and therapeutic management. Fetal Diagn Ther 14 (2): 102-5, 1999 Mar-Apr. [PUBMED Abstract]
Mazonakis M, Varveris H, Fasoulaki M, et al.: Radiotherapy of Hodgkin’s disease in early pregnancy: embryo dose measurements. Radiother Oncol 66 (3): 333-9, 2003. [PUBMED Abstract]
Fisher PM, Hancock BW: Hodgkin’s disease in the pregnant patient. Br J Hosp Med 56 (10): 529-32, 1996 Nov 20-Dec 10. [PUBMED Abstract]
Friedman E, Jones GW: Fetal outcome after maternal radiation treatment of supradiaphragmatic Hodgkin’s disease. CMAJ 149 (9): 1281-3, 1993. [PUBMED Abstract]
Woo SY, Fuller LM, Cundiff JH, et al.: Radiotherapy during pregnancy for clinical stages IA-IIA Hodgkin’s disease. Int J Radiat Oncol Biol Phys 23 (2): 407-12, 1992. [PUBMED Abstract]
Lishner M: Cancer in pregnancy. Ann Oncol 14 (Suppl 3): iii31-6, 2003. [PUBMED Abstract]
Cardonick E, Iacobucci A: Use of chemotherapy during human pregnancy. Lancet Oncol 5 (5): 283-91, 2004. [PUBMED Abstract]
Evens AM, Advani R, Press OW, et al.: Lymphoma occurring during pregnancy: antenatal therapy, complications, and maternal survival in a multicenter analysis. J Clin Oncol 31 (32): 4132-9, 2013. [PUBMED Abstract]
Pinnix CC, Osborne EM, Chihara D, et al.: Maternal and Fetal Outcomes After Therapy for Hodgkin or Non-Hodgkin Lymphoma Diagnosed During Pregnancy. JAMA Oncol 2 (8): 1065-9, 2016. [PUBMED Abstract]
Latest Updates to This Summary (01/13/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
This is a new summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of Hodgkin lymphoma during pregnancy. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Hodgkin Lymphoma Treatment During Pregnancy is:
Eric J. Seifter, MD (Johns Hopkins University)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
T-cell lymphoma can be divided into cutaneous T-cell lymphoma (CTCL), peripheral T-cell lymphoma (PTCL), and T-cell lymphoblastic lymphoma/acute lymphocytic leukemia (T-LBL/ALL).
T-LBL/ALL arises from very early T cells, often involves the thymus, and is more common in young adults. The lymphoma form is often treated similarly to the leukemia form. For more information, see Acute Lymphoblastic Leukemia Treatment.
PTCL originates from mature T cells. It usually arises from lymphoid tissues but can spread to other organs. Subsets of PTCL include anaplastic large cell lymphoma (ALCL), angioimmunoblastic T-cell lymphoma (AITL), extranodal natural killer/T-cell lymphoma (ENK/TCL), PTCL not otherwise specified (PTCL-NOS), enteropathy-associated T-cell lymphoma (EATL), hepatosplenic T-cell lymphoma (HSTCL), adult T-cell leukemia/lymphoma (ATL), T-cell prolymphocytic leukemia (T-PLL), and others.
Incidence and Mortality
T-cell lymphomas make up less than 15% of NHL cases. Most T-cell lymphoma subtypes are associated with worse outcomes than those of B-cell lymphomas.[1]
Anatomy
NHL usually originates in lymphoid tissues.
EnlargeThe lymph system is part of the body’s immune system and is made up of tissues and organs that help protect the body from infection and disease. These include the tonsils, adenoids (not shown), thymus, spleen, bone marrow, lymph vessels, and lymph nodes. Lymph tissue is also found in many other parts of the body, including the small intestine.
Prognosis and Survival
Prognosis in PTCL varies depending on subtype, stage, and other factors. In general, PTCL is associated with a poor prognosis, with a 5-year survival rate of approximately 30% to 40%.[2,3] While outcomes are better for patients with ALK-positive ALCL, with a median 5-year overall survival (OS) closer to 70% to 80%,[2–4] other subsets are associated with worse survival, such as ALK-negative ALCL, AITL, PTCL-NOS, HSTCL, EATL, and ENK/TCL.[5,6]
Unlike B-cell NHLs, which include both indolent and aggressive forms, most PTCLs are considered aggressive.[7] As with most other aggressive lymphomas, PTCLs are often curable with systemic therapy, though effective treatment options are more limited, particularly in the relapsed or refractory setting.[8,9]
Even though existing treatments cure a significant fraction of patients with lymphoma, numerous clinical trials that explore treatment improvements are in progress. If possible, patients can be included in these studies.
In addition to screening for HIV among patients with aggressive lymphomas, active hepatitis B or hepatitis C can be assessed before treatment with chemotherapy.[10,11] Patients with detectable hepatitis B virus (HBV) benefit from prophylaxis with entecavir.[12,13] Patients with a resolved HBV infection (defined as hepatitis B surface antigen-negative but hepatitis B core antibody-positive) are at risk of reactivation of HBV and require monitoring of HBV DNA. Prophylactic nucleoside therapy lowered HBV reactivation from 10.8% to 2.1% in a retrospective study of 326 patients.[14] Prophylaxis for herpes zoster with acyclovir or valacyclovir and prophylaxis for pneumocystis with trimethoprim/sulfamethoxazole or dapsone are usually given to patients receiving combination chemotherapy.
References
American Cancer Society: Types of T-cell lymphoma. American Cancer Society, 2018. Available online. Last accessed May 13, 2025.
Vose J, Armitage J, Weisenburger D, et al.: International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 26 (25): 4124-30, 2008. [PUBMED Abstract]
Ellin F, Landström J, Jerkeman M, et al.: Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: a study from the Swedish Lymphoma Registry. Blood 124 (10): 1570-7, 2014. [PUBMED Abstract]
Sibon D, Fournier M, Brière J, et al.: Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte trials. J Clin Oncol 30 (32): 3939-46, 2012. [PUBMED Abstract]
Petrich AM, Helenowski IB, Bryan LJ, et al.: Factors predicting survival in peripheral T-cell lymphoma in the USA: a population-based analysis of 8802 patients in the modern era. Br J Haematol 168 (5): 708-18, 2015. [PUBMED Abstract]
Foss FM, Horwitz SM, Civallero M, et al.: Incidence and outcomes of rare T cell lymphomas from the T Cell Project: hepatosplenic, enteropathy associated and peripheral gamma delta T cell lymphomas. Am J Hematol 95 (2): 151-155, 2020. [PUBMED Abstract]
Armitage JO: The aggressive peripheral T-cell lymphomas: 2017. Am J Hematol 92 (7): 706-715, 2017. [PUBMED Abstract]
Bellei M, Foss FM, Shustov AR, et al.: The outcome of peripheral T-cell lymphoma patients failing first-line therapy: a report from the prospective, International T-Cell Project. Haematologica 103 (7): 1191-1197, 2018. [PUBMED Abstract]
Lansigan F, Horwitz SM, Pinter-Brown LC, et al.: Outcomes for Relapsed and Refractory Peripheral T-Cell Lymphoma Patients after Front-Line Therapy from the COMPLETE Registry. Acta Haematol 143 (1): 40-50, 2020. [PUBMED Abstract]
Niitsu N, Hagiwara Y, Tanae K, et al.: Prospective analysis of hepatitis B virus reactivation in patients with diffuse large B-cell lymphoma after rituximab combination chemotherapy. J Clin Oncol 28 (34): 5097-100, 2010. [PUBMED Abstract]
Dong HJ, Ni LN, Sheng GF, et al.: Risk of hepatitis B virus (HBV) reactivation in non-Hodgkin lymphoma patients receiving rituximab-chemotherapy: a meta-analysis. J Clin Virol 57 (3): 209-14, 2013. [PUBMED Abstract]
Huang YH, Hsiao LT, Hong YC, et al.: Randomized controlled trial of entecavir prophylaxis for rituximab-associated hepatitis B virus reactivation in patients with lymphoma and resolved hepatitis B. J Clin Oncol 31 (22): 2765-72, 2013. [PUBMED Abstract]
Li H, Zhang HM, Chen LF, et al.: Prophylactic lamivudine to improve the outcome of HBsAg-positive lymphoma patients during chemotherapy: a systematic review and meta-analysis. Clin Res Hepatol Gastroenterol 39 (1): 80-92, 2015. [PUBMED Abstract]
Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019. [PUBMED Abstract]
Late Effects of Treatment of Peripheral T-Cell Non-Hodgkin Lymphoma
Late effects of treatment of non-Hodgkin lymphoma (NHL) have been observed. Impaired fertility may occur after exposure to alkylating agents.[1] For as many as three decades after diagnosis, patients are at a significantly elevated risk of developing second primary cancers, especially the following:[2–5]
Lung cancer.
Brain cancer.
Kidney cancer.
Bladder cancer.
Melanoma.
Hodgkin lymphoma.
Acute nonlymphocytic leukemia.
Left ventricular dysfunction was a significant late effect in long-term survivors of high-grade NHL who received more than 200 mg/m² of doxorubicin.[1,6]
Myelodysplastic syndrome and acute myelogenous leukemia are late complications of myeloablative therapy with autologous bone marrow or peripheral blood stem cell support, as well as conventional chemotherapy-containing alkylating agents.[3,7–14] Most of these patients show clonal hematopoiesis even before the transplant, suggesting that the hematologic injury usually occurs during induction or reinduction chemotherapy.[9,15,16] A series of 605 patients who received autologous bone marrow transplant (BMT) with cyclophosphamide and total-body radiation therapy (as conditioning) were followed for a median of 10 years. The incidence of a second malignancy was 21%, and 10% of those malignancies were solid tumors.[17]
A study of young women who received autologous BMT reported successful pregnancies with children born free of congenital abnormalities.[18] Late-occurring venous thromboembolism can occur after allogeneic or autologous BMT.[19]
Some patients have osteopenia or osteoporosis at the start of therapy; bone density may worsen after therapy for lymphoma.[20]
References
Haddy TB, Adde MA, McCalla J, et al.: Late effects in long-term survivors of high-grade non-Hodgkin’s lymphomas. J Clin Oncol 16 (6): 2070-9, 1998. [PUBMED Abstract]
Travis LB, Curtis RE, Glimelius B, et al.: Second cancers among long-term survivors of non-Hodgkin’s lymphoma. J Natl Cancer Inst 85 (23): 1932-7, 1993. [PUBMED Abstract]
Mudie NY, Swerdlow AJ, Higgins CD, et al.: Risk of second malignancy after non-Hodgkin’s lymphoma: a British Cohort Study. J Clin Oncol 24 (10): 1568-74, 2006. [PUBMED Abstract]
Hemminki K, Lenner P, Sundquist J, et al.: Risk of subsequent solid tumors after non-Hodgkin’s lymphoma: effect of diagnostic age and time since diagnosis. J Clin Oncol 26 (11): 1850-7, 2008. [PUBMED Abstract]
Major A, Smith DE, Ghosh D, et al.: Risk and subtypes of secondary primary malignancies in diffuse large B-cell lymphoma survivors change over time based on stage at diagnosis. Cancer 126 (1): 189-201, 2020. [PUBMED Abstract]
Moser EC, Noordijk EM, van Leeuwen FE, et al.: Long-term risk of cardiovascular disease after treatment for aggressive non-Hodgkin lymphoma. Blood 107 (7): 2912-9, 2006. [PUBMED Abstract]
Darrington DL, Vose JM, Anderson JR, et al.: Incidence and characterization of secondary myelodysplastic syndrome and acute myelogenous leukemia following high-dose chemoradiotherapy and autologous stem-cell transplantation for lymphoid malignancies. J Clin Oncol 12 (12): 2527-34, 1994. [PUBMED Abstract]
Stone RM, Neuberg D, Soiffer R, et al.: Myelodysplastic syndrome as a late complication following autologous bone marrow transplantation for non-Hodgkin’s lymphoma. J Clin Oncol 12 (12): 2535-42, 1994. [PUBMED Abstract]
Armitage JO, Carbone PP, Connors JM, et al.: Treatment-related myelodysplasia and acute leukemia in non-Hodgkin’s lymphoma patients. J Clin Oncol 21 (5): 897-906, 2003. [PUBMED Abstract]
André M, Mounier N, Leleu X, et al.: Second cancers and late toxicities after treatment of aggressive non-Hodgkin lymphoma with the ACVBP regimen: a GELA cohort study on 2837 patients. Blood 103 (4): 1222-8, 2004. [PUBMED Abstract]
Oddou S, Vey N, Viens P, et al.: Second neoplasms following high-dose chemotherapy and autologous stem cell transplantation for malignant lymphomas: a report of six cases in a cohort of 171 patients from a single institution. Leuk Lymphoma 31 (1-2): 187-94, 1998. [PUBMED Abstract]
Lenz G, Dreyling M, Schiegnitz E, et al.: Moderate increase of secondary hematologic malignancies after myeloablative radiochemotherapy and autologous stem-cell transplantation in patients with indolent lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group. J Clin Oncol 22 (24): 4926-33, 2004. [PUBMED Abstract]
McLaughlin P, Estey E, Glassman A, et al.: Myelodysplasia and acute myeloid leukemia following therapy for indolent lymphoma with fludarabine, mitoxantrone, and dexamethasone (FND) plus rituximab and interferon alpha. Blood 105 (12): 4573-5, 2005. [PUBMED Abstract]
Morton LM, Curtis RE, Linet MS, et al.: Second malignancy risks after non-Hodgkin’s lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 28 (33): 4935-44, 2010. [PUBMED Abstract]
Mach-Pascual S, Legare RD, Lu D, et al.: Predictive value of clonality assays in patients with non-Hodgkin’s lymphoma undergoing autologous bone marrow transplant: a single institution study. Blood 91 (12): 4496-503, 1998. [PUBMED Abstract]
Lillington DM, Micallef IN, Carpenter E, et al.: Detection of chromosome abnormalities pre-high-dose treatment in patients developing therapy-related myelodysplasia and secondary acute myelogenous leukemia after treatment for non-Hodgkin’s lymphoma. J Clin Oncol 19 (9): 2472-81, 2001. [PUBMED Abstract]
Brown JR, Yeckes H, Friedberg JW, et al.: Increasing incidence of late second malignancies after conditioning with cyclophosphamide and total-body irradiation and autologous bone marrow transplantation for non-Hodgkin’s lymphoma. J Clin Oncol 23 (10): 2208-14, 2005. [PUBMED Abstract]
Jackson GH, Wood A, Taylor PR, et al.: Early high dose chemotherapy intensification with autologous bone marrow transplantation in lymphoma associated with retention of fertility and normal pregnancies in females. Scotland and Newcastle Lymphoma Group, UK. Leuk Lymphoma 28 (1-2): 127-32, 1997. [PUBMED Abstract]
Gangaraju R, Chen Y, Hageman L, et al.: Risk of venous thromboembolism in patients with non-Hodgkin lymphoma surviving blood or marrow transplantation. Cancer 125 (24): 4498-4508, 2019. [PUBMED Abstract]
Westin JR, Thompson MA, Cataldo VD, et al.: Zoledronic acid for prevention of bone loss in patients receiving primary therapy for lymphomas: a prospective, randomized controlled phase III trial. Clin Lymphoma Myeloma Leuk 13 (2): 99-105, 2013. [PUBMED Abstract]
Cellular Classification of Peripheral T-Cell Non-Hodgkin Lymphoma
A pathologist should be consulted before a biopsy because some studies require special preparation of tissue (e.g., frozen tissue). Knowledge of cell surface markers and immunoglobulin and T-cell receptor gene rearrangements may help with diagnostic and therapeutic decisions. The clonal excess of light-chain immunoglobulin may differentiate malignant cells from reactive cells. Because the prognosis and the approach to treatment are influenced by histopathology, outside biopsy specimens should be carefully reviewed by a hematopathologist who is experienced in diagnosing lymphomas. Although lymph node biopsies are recommended whenever possible, sometimes immunophenotypic data are sufficient for diagnosis of lymphoma when fine-needle aspiration cytology or core needle biopsy is preferred.[1,2]
Current Classification Systems
Updated REAL/WHO classification
The World Health Organization (WHO) modification of the Revised European American Lymphoma (REAL) classification recognizes three major categories of lymphoid malignancies based on morphology and cell lineage: B-cell neoplasms, T-cell/natural killer (NK)-cell neoplasms, and Hodgkin lymphoma (HL). Both lymphomas and lymphoid leukemias are included in this classification because both solid and circulating phases are present in many lymphoid neoplasms and distinction between them is artificial. For example, B-cell chronic lymphocytic leukemia (CLL) and B-cell small lymphocytic lymphoma are simply different manifestations of the same neoplasm, as are lymphoblastic lymphomas and acute lymphocytic leukemias. Within the B-cell and T-cell categories, two subdivisions are recognized: precursor neoplasms, which correspond to the earliest stages of differentiation, and more mature differentiated neoplasms.[3,4]
Subtypes of Peripheral T-Cell Non-Hodgkin Lymphoma
Peripheral T-cell non-Hodgkin lymphoma includes the following subtypes, among others:
Anaplastic large cell lymphoma.
Nodal lymphomas of T follicular helper cell origin (including angioimmunoblastic T-cell lymphoma, follicular peripheral T-cell lymphoma, and nodal peripheral T-cell lymphoma with T follicular helper phenotype).
Peripheral T-cell lymphoma, not otherwise specified.
Extranodal NK/T-cell lymphoma.
Enteropathy-associated and monomorphic epitheliotropic intestinal T-cell lymphoma.
Hepatosplenic T-cell lymphoma.
Indolent T-cell lymphoma of the gastrointestinal tract.
Adult T-cell leukemia/lymphoma.
T-cell prolymphocytic leukemia.
Cutaneous T-cell lymphoma (including mycosis fungoides and Sézary syndrome, subcutaneous panniculitis-like T-cell lymphoma, primary cutaneous anaplastic large cell lymphoma, primary cutaneous gamma-delta T-cell lymphoma, and others). For more information, see Mycosis Fungoides and Other Cutaneous T-Cell Lymphomas Treatment.
Zeppa P, Marino G, Troncone G, et al.: Fine-needle cytology and flow cytometry immunophenotyping and subclassification of non-Hodgkin lymphoma: a critical review of 307 cases with technical suggestions. Cancer 102 (1): 55-65, 2004. [PUBMED Abstract]
Young NA, Al-Saleem T: Diagnosis of lymphoma by fine-needle aspiration cytology using the revised European-American classification of lymphoid neoplasms. Cancer 87 (6): 325-45, 1999. [PUBMED Abstract]
Pileri SA, Milani M, Fraternali-Orcioni G, et al.: From the R.E.A.L. Classification to the upcoming WHO scheme: a step toward universal categorization of lymphoma entities? Ann Oncol 9 (6): 607-12, 1998. [PUBMED Abstract]
Society for Hematopathology Program: Society for Hematopathology Program. Am J Surg Pathol 21 (1): 114-121, 1997.
Stage Information for Peripheral T-Cell Non-Hodgkin Lymphoma
Stage is important in selecting a treatment for patients with non-Hodgkin lymphoma (NHL). Chest and abdominal computed tomography (CT) scans are usually part of the staging evaluation for all patients with lymphoma. The staging system for NHL is similar to the staging system used for Hodgkin lymphoma (HL).
It is common for patients with NHL to have involvement of the following sites:
Noncontiguous lymph nodes.
Waldeyer ring.
Epitrochlear nodes.
Gastrointestinal tract.
Extranodal presentations. (A single extranodal site is occasionally the only site of involvement in patients with diffuse lymphoma.)
Bone marrow.
Liver (especially common in patients with low-grade lymphomas).
Cytological examination of cerebrospinal fluid may be positive in patients with aggressive NHL. Involvement of hilar and mediastinal lymph nodes is less common than in HL. Mediastinal adenopathy, however, is a prominent feature of lymphoblastic lymphoma and primary mediastinal B-cell lymphoma, entities primarily found in young adults.
Most patients with NHL present with advanced (stage III or stage IV) disease often identified by CT scans or biopsies of the bone marrow and other accessible sites of involvement. In a retrospective review of over 32,000 cases of lymphoma in France, up to 40% of diagnoses were made by core needle biopsy, and 60% were made by excisional biopsy.[1] After expert review, core needle biopsy provided a definite diagnosis in 92.3% of cases; excisional biopsy provided a definite diagnosis in 98.1% of cases (P < .0001). Laparoscopic biopsy or laparotomy is not required for staging but rarely may be necessary to establish a diagnosis or histological type.[2]
Positron emission tomography (PET) with fluorine F 18-fludeoxyglucose can be used for initial staging. It can also be used for follow-up after therapy as a supplement to CT scanning.[3] Multiple studies have demonstrated that interim PET scans after two to four cycles of therapy do not provide reliable prognostic information. A large cooperative group trial (ECOG-E344 [NCT00274924]) reported problems with interobserver reproducibility. Two prospective trials and one meta-analysis showed no differences in outcomes between PET-negative and PET-positive/biopsy-negative patients.[4–7]
In a retrospective study of 130 patients with diffuse large B-cell lymphoma, PET scanning identified all clinically important marrow involvement from lymphoma, and bone marrow biopsy did not upstage any patient’s lymphoma.[8] A retrospective study of 580 patients with follicular lymphoma from seven National Cancer Institute–sponsored trials showed no improvement in assessing response to therapy when bone marrow biopsy was added to radiological imaging.[9] The workup of NHL should include bone marrow biopsy when management would change (e.g., determining limited stage vs. advanced stage) or when evaluating cytopenias.
Staging Subclassification System
Lugano classification
The American Joint Committee on Cancer (AJCC) has adopted the Lugano classification to evaluate and stage lymphoma.[10] The Lugano classification system replaces the Ann Arbor classification system, which was adopted in 1971 at the Ann Arbor Conference,[11] with some modifications 18 years later from the Cotswolds meeting.[12,13]
Table 1. Lugano Classification for Hodgkin and Non-Hodgkin Lymphomaa
Stage
Stage Description
Illustration
CSF = cerebrospinal fluid; CT = computed tomography; DLBCL = diffuse large B-cell lymphoma; NHL = non-Hodgkin lymphoma.
aHodgkin and Non-Hodgkin Lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 937–58.
bStage II bulky may be considered either early or advanced stage based on lymphoma histology and prognostic factors.
cThe definition of disease bulk varies according to lymphoma histology. In the Lugano classification, bulk ln Hodgkin lymphoma is defined as a mass greater than one-third of the thoracic diameter on CT of the chest or a mass >10 cm. For NHL, the recommended definitions of bulk vary by lymphoma histology. In follicular lymphoma, 6 cm has been suggested based on the Follicular Lymphoma International Prognostic Index-2 and its validation. In DLBCL, cutoffs ranging from 5 cm to 10 cm have been used, although 10 cm is recommended.
Limited stage
I
Involvement of a single lymphatic site (i.e., nodal region, Waldeyer’s ring, thymus, or spleen).
Diffuse or disseminated involvement of one or more extralymphatic organs, with or without associated lymph node involvement; or noncontiguous extralymphatic organ involvement in conjunction with nodal stage II disease; or any extralymphatic organ involvement in nodal stage III disease. Stage IV includes any involvement of the CSF, bone marrow, liver, or multiple lung lesions (other than by direct extension in stage IIE disease).
Note: Hodgkin lymphoma uses A or B designation with stage group. A/B is no longer used in NHL.
Occasionally, specialized staging systems are used. The physician should be aware of the system used in a specific report.
The E designation is used when extranodal lymphoid malignancies arise in tissues separate from, but near, the major lymphatic aggregates. Stage IV refers to disease that is diffusely spread throughout an extranodal site, such as the liver. If pathological proof of involvement of one or more extralymphatic sites has been documented, the symbol for the site of involvement, followed by a plus sign (+), is listed.
Table 2. Notation to Identify Specific Sites
N = nodes
H = liver
L = lung
M = bone marrow
S = spleen
P = pleura
O = bone
D = skin
Current practice assigns a clinical stage based on the findings of the clinical evaluation and a pathological stage based on the findings from invasive procedures beyond the initial biopsy.
For example, on percutaneous biopsy, a patient with inguinal adenopathy and a positive lymphangiogram without systemic symptoms might have involvement of the liver and bone marrow. The precise stage of such a patient would be clinical stage IIA, pathological stage IVA(H+)(M+).
Several other factors that are not included in the above staging system are important for the staging and prognosis of patients with NHL. These factors include the following:
Age.
Performance status (PS).
Tumor size.
Lactate dehydrogenase (LDH) values.
The number of extranodal sites.
The National Comprehensive Cancer Network International Prognostic Index (IPI) for aggressive NHL (diffuse large cell lymphoma) identifies the following five significant risk factors prognostic of overall survival (OS) and their associated risk scores:[14]
Age.
<40 years: 0.
41–60 years: 1.
61–75 years: 2.
>75 years: 3.
Stage III/IV: 1.
Performance status (PS) 2/3/4: 1.
Serum lactate dehydrogenase (LDH).
Normalized: 0.
>1x–3x: 1.
>3x: 2.
Number of extranodal sites ≥2: 1.
Risk scores:
Low (0 or 1): 5-year OS rate, 96%; progression-free survival (PFS) rate, 91%.
Low intermediate (2 or 3): 5-year OS rate, 82%; PFS rate, 74%.
High intermediate (4 or 5): 5-year OS rate, 64%; PFS rate, 51%.
High (>6): 5-year OS rate, 33%; PFS rate, 30%.
References
Syrykh C, Chaouat C, Poullot E, et al.: Lymph node excisions provide more precise lymphoma diagnoses than core biopsies: a French Lymphopath network survey. Blood 140 (24): 2573-2583, 2022. [PUBMED Abstract]
Mann GB, Conlon KC, LaQuaglia M, et al.: Emerging role of laparoscopy in the diagnosis of lymphoma. J Clin Oncol 16 (5): 1909-15, 1998. [PUBMED Abstract]
Barrington SF, Mikhaeel NG, Kostakoglu L, et al.: Role of imaging in the staging and response assessment of lymphoma: consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 32 (27): 3048-58, 2014. [PUBMED Abstract]
Horning SJ, Juweid ME, Schöder H, et al.: Interim positron emission tomography scans in diffuse large B-cell lymphoma: an independent expert nuclear medicine evaluation of the Eastern Cooperative Oncology Group E3404 study. Blood 115 (4): 775-7; quiz 918, 2010. [PUBMED Abstract]
Moskowitz CH, Schöder H, Teruya-Feldstein J, et al.: Risk-adapted dose-dense immunochemotherapy determined by interim FDG-PET in Advanced-stage diffuse large B-Cell lymphoma. J Clin Oncol 28 (11): 1896-903, 2010. [PUBMED Abstract]
Pregno P, Chiappella A, Bellò M, et al.: Interim 18-FDG-PET/CT failed to predict the outcome in diffuse large B-cell lymphoma patients treated at the diagnosis with rituximab-CHOP. Blood 119 (9): 2066-73, 2012. [PUBMED Abstract]
Sun N, Zhao J, Qiao W, et al.: Predictive value of interim PET/CT in DLBCL treated with R-CHOP: meta-analysis. Biomed Res Int 2015: 648572, 2015. [PUBMED Abstract]
Khan AB, Barrington SF, Mikhaeel NG, et al.: PET-CT staging of DLBCL accurately identifies and provides new insight into the clinical significance of bone marrow involvement. Blood 122 (1): 61-7, 2013. [PUBMED Abstract]
Rutherford SC, Yin J, Pederson L, et al.: Relevance of Bone Marrow Biopsies for Response Assessment in US National Cancer Institute National Clinical Trials Network Follicular Lymphoma Clinical Trials. J Clin Oncol 41 (2): 336-342, 2023. [PUBMED Abstract]
Hodgkin and non-Hodgkin lymphoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 937–58.
Carbone PP, Kaplan HS, Musshoff K, et al.: Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res 31 (11): 1860-1, 1971. [PUBMED Abstract]
Lister TA, Crowther D, Sutcliffe SB, et al.: Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol 7 (11): 1630-6, 1989. [PUBMED Abstract]
National Cancer Institute sponsored study of classifications of non-Hodgkin’s lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin’s Lymphoma Pathologic Classification Project. Cancer 49 (10): 2112-35, 1982. [PUBMED Abstract]
Zhou Z, Sehn LH, Rademaker AW, et al.: An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123 (6): 837-42, 2014. [PUBMED Abstract]
Treatment of Anaplastic Large Cell Lymphoma
Anaplastic large cell lymphoma (ALCL) is a peripheral T-cell lymphoma associated with the CD30 antigen. The translocation of chromosomes 2 and 5 creates a unique fusion protein with a nucleophosmin–anaplastic lymphoma kinase (ALK).[1,2] Patients whose lymphomas express ALK by immunohistochemistry are usually younger and may have systemic symptoms, extranodal disease, and advanced-stage disease. However, they have a more favorable survival rate than patients with ALK-negative disease.[3,4] ALK-negative ALCL has been further characterized by DUSP22 chromosomal rearrangements and the presence of TP63 pathogenic variants. While DUSP22 rearrangements are associated with improved prognosis, TP63 pathogenic variants are associated with poorer outcomes.[5]
Treatment Options for Anaplastic Large Cell Lymphoma
A prospective randomized trial included 452 patients with CD30-positive T-cell lymphoma (CD30 expression >10%). Of these patients, 70% had ALCL (22% with ALK-positive disease and 48% with ALK-negative disease). The trial compared the previously used standard regimen, CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), with brentuximab vedotin (an anti-CD30 monoclonal antibody conjugated to a cytotoxic agent) combined with cyclophosphamide, doxorubicin, and prednisone (A+CHP regimen).[6]
With a median follow-up of 47.6 months, the 5-year overall survival (OS) rates were 70.1% (95% confidence interval [CI], 63.3%–75.9%) for patients who received A+CHP and 61.0% (95% CI, 54.0%–67.3%) for patients who received CHOP (hazard ratio [HR], 0.72; 95% CI, 0.53–0.99).[7][Level of evidence A1]
The 5-year progression-free survival (PFS) rates were 51.4% (95% CI, 42.8%–59.4%) for patients who received A+CHP and 43.0% (95% CI, 35.8%–50.0%) for patients who received CHOP (HR, 0.70; 95% CI, 0.53–0.91).
This established A+CHP as a new option for patients with ALCL or other CD30-positive T-cell lymphomas, such as angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, not otherwise specified.
For patients with relapsed disease, anecdotal responses have been reported for brentuximab vedotin,[8–11] belinostat,[12] romidepsin,[13] and pralatrexate.[14][Level of evidence C3]
In a phase II study (NCT00866047), 66% of 58 patients attained a complete response with brentuximab vedotin.[11]
At a median follow-up of 58 months, the 5-year PFS rate was 57% (95% CI, 41%–74%), and the 5-year OS rate was 79% (95% CI, 65%–92%). Of the patients achieving a complete response, 42% underwent hematopoietic stem cell transplant (SCT).[11][Level of evidence C3]
In a retrospective review, 39 patients with relapsed disease had a 3-year PFS rate of 50% after autologous or allogeneic SCT.[15][Level of evidence C2]
A retrospective review of 84 patients with ALK-negative ALCL suggested a survival benefit with autologous SCT. This hypothesis requires confirmation in a randomized prospective trial.[16][Level of evidence C3]
A retrospective study included 182 patients with relapsed or refractory ALCL (23% ALK-positive, 21% ALK-negative, and 56% ALK-unknown) who underwent allogeneic SCT.[17]
The 5-year PFS rate was 41% (95% CI, 34%–49%), and the 5-year OS rate was 41% (95% CI, 49%–64%).[17][Level of evidence C3]
On multivariate analysis, African American race (HR, 2.7; 95% CI, 1.6–4.8; P < .001) and refractory disease at time of allogeneic SCT (HR, 3.2; 95% CI, 1.6–6.2; P < .001) were predictive factors for inferior OS.
Despite ALK positivity being a favorable prognostic factor, outcomes after allogeneic SCT in this study did not vary significantly according to ALK status.
ALCL in children is usually characterized by systemic and cutaneous disease and has high response rates and good OS with doxorubicin-based combination chemotherapy.[18] The ALK inhibitor crizotinib has been combined with chemotherapy for previously untreated pediatric patients, and crizotinib has been used to control disease in multiply relapsed pediatric patients.[19,20] Crizotinib is associated with a high risk (around 25%) of thromboembolism, especially pulmonary embolism, and prophylaxis is recommended. There are no reports supporting the use of crizotinib in adults.
Breast Implant–Associated Anaplastic Large Cell Lymphoma
Patients with breast implant–associated ALCL may do well without chemotherapy after capsulectomy and implant removal if the disease is confined to the fibrous capsule, and no associated mass or lymphadenopathy is present.[21–24] Most patients with breast implant–associated ALCL have a characteristic deletion at 20Q13.13 that may help to diagnostically distinguish it from cutaneous or systemic ALCL.[25]
Primary cutaneous ALCL is a distinct entity that is typically ALK-negative and has a very indolent/low-grade clinical course. Breast implant–associated ALCL almost always occurs with implants that have a textured surface (so-called macrotextured implants) that helps adhere to the breast. Warnings, suspensions, and bans of macrotextured implants have occurred worldwide. The risk of breast implant–associated ALCL is approximately 1 in 12,000 people, but this risk may rise with further follow-up.[26]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Bai RY, Ouyang T, Miething C, et al.: Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood 96 (13): 4319-27, 2000. [PUBMED Abstract]
Hapgood G, Savage KJ: The biology and management of systemic anaplastic large cell lymphoma. Blood 126 (1): 17-25, 2015. [PUBMED Abstract]
Gascoyne RD, Aoun P, Wu D, et al.: Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood 93 (11): 3913-21, 1999. [PUBMED Abstract]
Sibon D, Fournier M, Brière J, et al.: Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte trials. J Clin Oncol 30 (32): 3939-46, 2012. [PUBMED Abstract]
Onaindia A, de Villambrosía SG, Prieto-Torres L, et al.: DUSP22-rearranged anaplastic lymphomas are characterized by specific morphological features and a lack of cytotoxic and JAK/STAT surrogate markers. Haematologica 104 (4): e158-e162, 2019. [PUBMED Abstract]
Horwitz S, O’Connor OA, Pro B, et al.: Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393 (10168): 229-240, 2019. [PUBMED Abstract]
Horwitz S, O’Connor OA, Pro B, et al.: The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol 33 (3): 288-298, 2022. [PUBMED Abstract]
Younes A, Bartlett NL, Leonard JP, et al.: Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 363 (19): 1812-21, 2010. [PUBMED Abstract]
Pro B, Advani R, Brice P, et al.: Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 30 (18): 2190-6, 2012. [PUBMED Abstract]
Prince HM, Kim YH, Horwitz SM, et al.: Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet 390 (10094): 555-566, 2017. [PUBMED Abstract]
Pro B, Advani R, Brice P, et al.: Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 130 (25): 2709-2717, 2017. [PUBMED Abstract]
O’Connor OA, Horwitz S, Masszi T, et al.: Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 33 (23): 2492-9, 2015. [PUBMED Abstract]
Coiffier B, Pro B, Prince HM, et al.: Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30 (6): 631-6, 2012. [PUBMED Abstract]
O’Connor OA, Horwitz S, Hamlin P, et al.: Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol 27 (26): 4357-64, 2009. [PUBMED Abstract]
Smith SM, Burns LJ, van Besien K, et al.: Hematopoietic cell transplantation for systemic mature T-cell non-Hodgkin lymphoma. J Clin Oncol 31 (25): 3100-9, 2013. [PUBMED Abstract]
Brink M, Meeuwes FO, van der Poel MWM, et al.: Impact of etoposide and ASCT on survival among patients aged <65 years with stage II to IV PTCL: a population-based cohort study. Blood 140 (9): 1009-1019, 2022. [PUBMED Abstract]
Furqan F, Ahn KW, Chen Y, et al.: Allogeneic haematopoietic cell transplant in patients with relapsed/refractory anaplastic large cell lymphoma. Br J Haematol 200 (1): 54-63, 2023. [PUBMED Abstract]
Seidemann K, Tiemann M, Schrappe M, et al.: Short-pulse B-non-Hodgkin lymphoma-type chemotherapy is efficacious treatment for pediatric anaplastic large cell lymphoma: a report of the Berlin-Frankfurt-Münster Group Trial NHL-BFM 90. Blood 97 (12): 3699-706, 2001. [PUBMED Abstract]
Lowe EJ, Reilly AF, Lim MS, et al.: Crizotinib in Combination With Chemotherapy for Pediatric Patients With ALK+ Anaplastic Large-Cell Lymphoma: The Results of Children’s Oncology Group Trial ANHL12P1. J Clin Oncol 41 (11): 2043-2053, 2023. [PUBMED Abstract]
Mossé YP, Voss SD, Lim MS, et al.: Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J Clin Oncol 35 (28): 3215-3221, 2017. [PUBMED Abstract]
Miranda RN, Aladily TN, Prince HM, et al.: Breast implant-associated anaplastic large-cell lymphoma: long-term follow-up of 60 patients. J Clin Oncol 32 (2): 114-20, 2014. [PUBMED Abstract]
Clemens MW, Medeiros LJ, Butler CE, et al.: Complete Surgical Excision Is Essential for the Management of Patients With Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J Clin Oncol 34 (2): 160-8, 2016. [PUBMED Abstract]
Mehta-Shah N, Clemens MW, Horwitz SM: How I treat breast implant-associated anaplastic large cell lymphoma. Blood 132 (18): 1889-1898, 2018. [PUBMED Abstract]
Jaffe ES, Ashar BS, Clemens MW, et al.: Best Practices Guideline for the Pathologic Diagnosis of Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J Clin Oncol 38 (10): 1102-1111, 2020. [PUBMED Abstract]
Los-de Vries GT, de Boer M, van Dijk E, et al.: Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lymphoma. Blood 136 (25): 2927-2932, 2020. [PUBMED Abstract]
Kinslow CJ, Kim A, Sanchez GI, et al.: Incidence of Anaplastic Large-Cell Lymphoma of the Breast in the US, 2000 to 2018. JAMA Oncol 8 (9): 1354-1356, 2022. [PUBMED Abstract]
Treatment of Nodal Lymphomas of T Follicular Helper Cell Origin
The 2016 World Health Organization (WHO) classification recognized nodal lymphomas of T follicular helper (TFH) cell origin as a distinct subset of peripheral T-cell lymphoma (PTCL). Unified by a CD4+ TFH cell origin, this subset includes:[1]
Angioimmunoblastic T-cell lymphoma (AITL or ATCL).
Follicular peripheral T-cell lymphoma (F-PTCL).
Nodal peripheral T-cell lymphoma with follicular helper phenotype (nodal PTCL with TFH phenotype).
AITL is the most common of these entities and is the second-most common subtype of PTCL.[2–5] Patients often present with profound lymphadenopathy, fever, night sweats, weight loss, skin rash, a positive Coombs test, and polyclonal hypergammaglobulinemia.[6] Opportunistic infections are frequent because of an underlying immune deficiency. B-cell Epstein-Barr virus genomes are detected in most affected patients.[7] For more information about weight loss, see Nutrition in Cancer Care and for more information about skin rash, see Pruritus.
The remaining subtypes of nodal lymphomas of TFH cell origin, including F-PTCL and nodal PTCL with TFH phenotype, are derived from the same cell of origin. However, unlike AITL, these subtypes are not associated with hypervascularity on nodal biopsy and often lack the hyperinflammatory symptoms characteristic of AITL.[1]
Treatment Options for Nodal Lymphomas of T Follicular Helper Cell Origin
Doxorubicin-based combination chemotherapy, such as the CHO(E)P regimen (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone), is commonly used for patients with AITL and other nodal lymphomas of TFH cell origin.[2,5] For CD30-positive cases, brentuximab vedotin combined with cyclophosphamide, doxorubicin, and prednisone is the proposed standard of care.[8][Level of evidence C3] Patients with AITL were included in a clinical trial involving mostly patients with anaplastic large cell lymphoma. A benefit for this smaller AITL subgroup cannot be established.[8,9][Level of evidence C3] For more information, see the Treatment of Anaplastic Large Cell Lymphoma section.
The International Peripheral T-Cell Lymphoma Project involving 22 international centers identified 243 patients with AITL. The 5-year overall survival rate was 33%, and the failure-free survival rate was 18%.[10] Myeloablative chemotherapy and radiation therapy with autologous or allogeneic peripheral stem cell support has been described in anecdotal reports.[11–16][Level of evidence C3] Anecdotal responses have been reported for patients who received cyclosporine,[17] pralatrexate,[18] bendamustine,[19] belinostat,[20] the histone deacetylase inhibitor romidepsin, hypomethylating agents (HMAs),[21,22] and brentuximab vedotin (even if there is little or no CD30 expression on the lymphoma).[23,24][Level of evidence C3] Occasional spontaneous remissions and protracted responses to steroids only have been reported. Given increased frequency of pathogenic variants in genes that regulate epigenetic modification—such as TET2, IDH2, and DNMT3A—there is increased interest in therapies targeting these aberrations with HMAs and histone deacetylase inhibitors with anecdotal reports of improved efficacy compared with other subsets of PTCL.[21,22,25][Level of evidence C3] Further studies are needed to better characterize the efficacy of this approach.[21,22,25]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Yoon SE, Cho J, Kim YJ, et al.: Comprehensive analysis of clinical, pathological, and genomic characteristics of follicular helper T-cell derived lymphomas. Exp Hematol Oncol 10 (1): 33, 2021. [PUBMED Abstract]
Siegert W, Agthe A, Griesser H, et al.: Treatment of angioimmunoblastic lymphadenopathy (AILD)-type T-cell lymphoma using prednisone with or without the COPBLAM/IMVP-16 regimen. A multicenter study. Kiel Lymphoma Study Group. Ann Intern Med 117 (5): 364-70, 1992. [PUBMED Abstract]
Jaffe ES: Angioimmunoblastic T-cell lymphoma: new insights, but the clinical challenge remains. Ann Oncol 6 (7): 631-2, 1995. [PUBMED Abstract]
Siegert W, Nerl C, Agthe A, et al.: Angioimmunoblastic lymphadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group. Ann Oncol 6 (7): 659-64, 1995. [PUBMED Abstract]
Bräuninger A, Spieker T, Willenbrock K, et al.: Survival and clonal expansion of mutating “forbidden” (immunoglobulin receptor-deficient) epstein-barr virus-infected b cells in angioimmunoblastic t cell lymphoma. J Exp Med 194 (7): 927-40, 2001. [PUBMED Abstract]
Horwitz S, O’Connor OA, Pro B, et al.: Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393 (10168): 229-240, 2019. [PUBMED Abstract]
Horwitz S, O’Connor OA, Pro B, et al.: The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol 33 (3): 288-298, 2022. [PUBMED Abstract]
Federico M, Rudiger T, Bellei M, et al.: Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the international peripheral T-cell lymphoma project. J Clin Oncol 31 (2): 240-6, 2013. [PUBMED Abstract]
Reimer P, Rüdiger T, Geissinger E, et al.: Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: results of a prospective multicenter study. J Clin Oncol 27 (1): 106-13, 2009. [PUBMED Abstract]
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008. [PUBMED Abstract]
Kyriakou C, Canals C, Finke J, et al.: Allogeneic stem cell transplantation is able to induce long-term remissions in angioimmunoblastic T-cell lymphoma: a retrospective study from the lymphoma working party of the European group for blood and marrow transplantation. J Clin Oncol 27 (24): 3951-8, 2009. [PUBMED Abstract]
Park SI, Horwitz SM, Foss FM, et al.: The role of autologous stem cell transplantation in patients with nodal peripheral T-cell lymphomas in first complete remission: Report from COMPLETE, a prospective, multicenter cohort study. Cancer 125 (9): 1507-1517, 2019. [PUBMED Abstract]
Brink M, Meeuwes FO, van der Poel MWM, et al.: Impact of etoposide and ASCT on survival among patients aged <65 years with stage II to IV PTCL: a population-based cohort study. Blood 140 (9): 1009-1019, 2022. [PUBMED Abstract]
Los-de Vries GT, de Boer M, van Dijk E, et al.: Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lymphoma. Blood 136 (25): 2927-2932, 2020. [PUBMED Abstract]
Advani R, Horwitz S, Zelenetz A, et al.: Angioimmunoblastic T cell lymphoma: treatment experience with cyclosporine. Leuk Lymphoma 48 (3): 521-5, 2007. [PUBMED Abstract]
Amengual JE, Lichtenstein R, Lue J, et al.: A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 131 (4): 397-407, 2018. [PUBMED Abstract]
Damaj G, Gressin R, Bouabdallah K, et al.: Results from a prospective, open-label, phase II trial of bendamustine in refractory or relapsed T-cell lymphomas: the BENTLY trial. J Clin Oncol 31 (1): 104-10, 2013. [PUBMED Abstract]
O’Connor OA, Horwitz S, Masszi T, et al.: Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 33 (23): 2492-9, 2015. [PUBMED Abstract]
Wong J, Gruber E, Maher B, et al.: Integrated clinical and genomic evaluation of guadecitabine (SGI-110) in peripheral T-cell lymphoma. Leukemia 36 (6): 1654-1665, 2022. [PUBMED Abstract]
Lemonnier F, Dupuis J, Sujobert P, et al.: Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood 132 (21): 2305-2309, 2018. [PUBMED Abstract]
Coiffier B, Pro B, Prince HM, et al.: Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30 (6): 631-6, 2012. [PUBMED Abstract]
Fanale MA, Horwitz SM, Forero-Torres A, et al.: Five-year outcomes for frontline brentuximab vedotin with CHP for CD30-expressing peripheral T-cell lymphomas. Blood 131 (19): 2120-2124, 2018. [PUBMED Abstract]
Pro B, Horwitz SM, Prince HM, et al.: Romidepsin induces durable responses in patients with relapsed or refractory angioimmunoblastic T-cell lymphoma. Hematol Oncol 35 (4): 914-917, 2017. [PUBMED Abstract]
Treatment of Peripheral T-Cell Lymphoma, Not Otherwise Specified
Patients with peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS) have diffuse large cell or diffuse mixed lymphoma that expresses a cell surface phenotype of a postthymic (or peripheral) T-cell expressing either CD4 or, less often, CD8.[1] PTCL-NOS encompasses a group of heterogeneous nodal T-cell lymphomas that will require future delineation.[2,3] Gene expression profiling studies have identified two distinct subtypes of PTCL-NOS based on the transcription factors GATA3 and TBX21. Early investigation and anecdotal reports suggest these subtypes may carry important prognostic implications and could predict response to certain therapies.[4] Additional studies are needed for further characterization of their clinical relevance.
Prognosis
Most investigators report worse response and survival rates for patients with PTCL-NOS than for patients with comparably staged B-cell aggressive lymphomas.[3,5] Most patients present with multiple adverse prognostic factors (i.e., older age, stage IV, multiple extranodal sites, and elevated lactate dehydrogenase), and these patients have low (<20%) failure-free survival and overall survival (OS) rates at 5 years.[3,5] As with other lymphomas (e.g., diffuse large B-cell lymphoma [DLBCL] or follicular lymphoma), event-free survival (EFS) at 24 months predicts a 5-year OS rate of 78%.[6]
Treatment Options for Peripheral T-Cell Lymphoma, Not Otherwise Specified
Therapy involves doxorubicin-based combination chemotherapy such as cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or CHO(E)P (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone). Doses are the same as those used for DLBCL.[7] For CD30-positive cases, brentuximab vedotin combined with cyclophosphamide, doxorubicin, and prednisone is the proposed standard of care.[8][Level of evidence C3] Patients with PTCL-NOS were included in the clinical trial involving mostly patients with anaplastic large cell lymphoma. A benefit for this smaller PTCL-NOS subgroup cannot be established.[8,9][Level of evidence C3] For more information, see the Treatment of Anaplastic Large Cell Lymphoma section.
For patients with early-stage disease, anecdotal retrospective series disagree on the value of consolidative radiation therapy after combination chemotherapy.[10][Level of evidence C3] Consolidation therapy using high-dose chemotherapy with autologous or allogeneic hematopoietic stem cell transplant (SCT) has been given to patients with advanced-stage PTCL after induction therapy in multiple phase II or retrospective trials. Evidence for this approach is anecdotal.[11–18][Level of evidence C3]
Evidence (CHOP, CHO[E]P, or other options for relapsing disease):
A randomized prospective trial included 104 patients younger than 61 years with stage II, III, or IV PTCL (excluding ALK-positive anaplastic large cell lymphoma). Patients received either autologous SCT or allogeneic SCT as consolidation therapy after induction with CHO(E)P followed by DHAP (dexamethasone, cytarabine, and cisplatin).[19][Level of evidence C3]
With a median follow-up of 42 months, the 3-year EFS rate was 43% for patients who received allogeneic SCT and 38% for patients who received autologous SCT.
The 3-year OS rate was 57% for patients who received allogeneic SCT and 70% for patients who received autologous SCT (P = nonsignificant).
None of the 21 responding patients who proceeded to allogeneic SCT relapsed, and 36% of patients who proceeded to autologous SCT relapsed.
Eight of 26 patients (31%) who received allogeneic SCT died of graft-versus-host disease, and none of the 41 patients who received autologous SCT died of toxicity.
The benefit of graft-versus-lymphoma effect was negated by increased transplant-related mortality.
In a prospective trial of 109 evaluable patients with relapsing disease, treatment with pralatrexate resulted in a 30% response rate and a median 10-month duration of response.[20,21][Level of evidence C3]
Similar response rates were seen in 130 evaluable patients with relapsing disease who received romidepsin in a prospective trial.[21][Level of evidence C3]
Anecdotal responses have been seen with a combination of pralatrexate and romidepsin,[22] single-agent bendamustine,[23] belinostat,[24] and brentuximab vedotin (even if there is little or no CD30 expression on the lymphoma).[25][Level of evidence C3]
Incorporation of these new agents with CHOP chemotherapy is under clinical evaluation.[3,8]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Rüdiger T, Weisenburger DD, Anderson JR, et al.: Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkin’s Lymphoma Classification Project. Ann Oncol 13 (1): 140-9, 2002. [PUBMED Abstract]
Weisenburger DD, Savage KJ, Harris NL, et al.: Peripheral T-cell lymphoma, not otherwise specified: a report of 340 cases from the International Peripheral T-cell Lymphoma Project. Blood 117 (12): 3402-8, 2011. [PUBMED Abstract]
Amador C, Greiner TC, Heavican TB, et al.: Reproducing the molecular subclassification of peripheral T-cell lymphoma-NOS by immunohistochemistry. Blood 134 (24): 2159-2170, 2019. [PUBMED Abstract]
Sonnen R, Schmidt WP, Müller-Hermelink HK, et al.: The International Prognostic Index determines the outcome of patients with nodal mature T-cell lymphomas. Br J Haematol 129 (3): 366-72, 2005. [PUBMED Abstract]
Maurer MJ, Ellin F, Srour L, et al.: International Assessment of Event-Free Survival at 24 Months and Subsequent Survival in Peripheral T-Cell Lymphoma. J Clin Oncol 35 (36): 4019-4026, 2017. [PUBMED Abstract]
Carson KR, Horwitz SM, Pinter-Brown LC, et al.: A prospective cohort study of patients with peripheral T-cell lymphoma in the United States. Cancer 123 (7): 1174-1183, 2017. [PUBMED Abstract]
Horwitz S, O’Connor OA, Pro B, et al.: Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393 (10168): 229-240, 2019. [PUBMED Abstract]
Horwitz S, O’Connor OA, Pro B, et al.: The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol 33 (3): 288-298, 2022. [PUBMED Abstract]
Briski R, Feldman AL, Bailey NG, et al.: Survival in patients with limited-stage peripheral T-cell lymphomas. Leuk Lymphoma 56 (6): 1665-70, 2015. [PUBMED Abstract]
Rodriguez J, Munsell M, Yazji S, et al.: Impact of high-dose chemotherapy on peripheral T-cell lymphomas. J Clin Oncol 19 (17): 3766-70, 2001. [PUBMED Abstract]
Reimer P, Rüdiger T, Geissinger E, et al.: Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: results of a prospective multicenter study. J Clin Oncol 27 (1): 106-13, 2009. [PUBMED Abstract]
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008. [PUBMED Abstract]
Park SI, Horwitz SM, Foss FM, et al.: The role of autologous stem cell transplantation in patients with nodal peripheral T-cell lymphomas in first complete remission: Report from COMPLETE, a prospective, multicenter cohort study. Cancer 125 (9): 1507-1517, 2019. [PUBMED Abstract]
Brink M, Meeuwes FO, van der Poel MWM, et al.: Impact of etoposide and ASCT on survival among patients aged <65 years with stage II to IV PTCL: a population-based cohort study. Blood 140 (9): 1009-1019, 2022. [PUBMED Abstract]
Los-de Vries GT, de Boer M, van Dijk E, et al.: Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lymphoma. Blood 136 (25): 2927-2932, 2020. [PUBMED Abstract]
Schmitz N, Truemper L, Bouabdallah K, et al.: A randomized phase 3 trial of autologous vs allogeneic transplantation as part of first-line therapy in poor-risk peripheral T-NHL. Blood 137 (19): 2646-2656, 2021. [PUBMED Abstract]
O’Connor OA, Pro B, Pinter-Brown L, et al.: Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 29 (9): 1182-9, 2011. [PUBMED Abstract]
Coiffier B, Pro B, Prince HM, et al.: Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30 (6): 631-6, 2012. [PUBMED Abstract]
Amengual JE, Lichtenstein R, Lue J, et al.: A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 131 (4): 397-407, 2018. [PUBMED Abstract]
Damaj G, Gressin R, Bouabdallah K, et al.: Results from a prospective, open-label, phase II trial of bendamustine in refractory or relapsed T-cell lymphomas: the BENTLY trial. J Clin Oncol 31 (1): 104-10, 2013. [PUBMED Abstract]
O’Connor OA, Horwitz S, Masszi T, et al.: Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 33 (23): 2492-9, 2015. [PUBMED Abstract]
Fanale MA, Horwitz SM, Forero-Torres A, et al.: Five-year outcomes for frontline brentuximab vedotin with CHP for CD30-expressing peripheral T-cell lymphomas. Blood 131 (19): 2120-2124, 2018. [PUBMED Abstract]
Treatment of Extranodal Natural Killer/T-Cell Lymphoma
Extranodal natural killer (NK)/T-cell lymphoma, nasal type, is an aggressive lymphoma marked by extensive necrosis and angioinvasion, most often presenting in extranodal sites, in particular the nasal or paranasal sinus region.[1] Other extranodal sites include the palate, trachea, skin, and gastrointestinal tract. Hemophagocytic syndrome may also occur; historically, these tumors were considered part of lethal midline granuloma.[2] Tumor cells are typically NK cells with immunophenotyping showing CD3 and CD56 expression. In nearly all cases, Epstein-Barr virus (EBV) is detectable in the tumor cells. Aggressive NK cell leukemia (ANKL) is a separate but related diagnosis, which was previously recognized as a subset of large granular lymphocyte (LGL) leukemia, and is characterized by mature NK cell neoplastic involvement of the blood or marrow involvement with a particularly aggressive course and poor prognosis.[3] A benign NK-cell enteropathy (EBV negative) on endoscopic biopsy can be distinguished from NK/T-cell lymphoma.[3] Extranodal NK-cell lymphoma (ENKL), nasal type is rare in Western countries and more commonly seen in Asia, where it represents 3% to 8% of all lymphoma cases. Most cases present with nasal involvement and localized disease. Both extranasal and advanced-stage (stages III–IV) disease are associated with poor prognosis.[4,5]
Treatment Options for Extranodal Natural Killer/T-Cell Lymphoma
The management of localized ENKL with nasal involvement involves combined modality therapy with chemotherapy and radiation therapy for those fit for chemotherapy or radiation therapy alone for those unfit for chemotherapy. The management of nonlocalized nasal ENKL and all cases of extranasal ENKL involves combination chemotherapy with or without radiation therapy.[6] Because ENKL frequently expresses P-glycoprotein which confers multidrug resistance and reduces the efficacy of traditional anthracycline-based regimens, asparaginase is typically incorporated into the chemotherapy regimens.[7,8] Various asparaginase-based combination chemotherapy regimens have been used.
A phase II study included 28 patients with newly diagnosed stage IV or relapsed or refractory ENKL. Patients received steroids, methotrexate, ifosfamide, [L-]asparaginase, and etoposide (SMILE).[9]
The overall response rate after two cycles was 79% (95% confidence interval [CI], 65%–89%), and the complete response rate was 45%. The 1-year overall survival (OS) rate was 55% (95% CI, 38%–69%).
Grade 4 neutropenia was observed in 92% of patients, and grade 3 or 4 infections were seen in 61% of patients.[9]
A modification to the SMILE regimen (mSMILE) incorporating pegylated asparaginase has demonstrated reduced toxicity.[10]
Despite no head-to-head studies, this regimen is used more often in clinical practice, in patients with advanced-stage disease, or in combination with radiation therapy for patients with localized disease. Other asparaginase-based regimens which have been studied include gemcitabine, oxaliplatin, and pegaspargase (P-GEMOX);[10] gemcitabine, etoposide, pegaspargase, and dexamethasone (GELAD);[11] and dexamethasone, cisplatin, gemcitabine, and pegaspargase (DDGP).[12]For patients who may not tolerate or who have reactions to asparaginase, a nonasparaginase regimen may be given with concurrent radiation therapy.[13][Level of evidence C3]
A retrospective study (NCT02733458) included 35 patients with newly diagnosed stage III to IV, relapsed, or refractory ENKL. Patients received P-GEMOX.[10]
The overall response rate was 80.0%, and the complete response rate was 51.4%. The 3-year progression-free survival (PFS) rate was 38.6%, and the 3-year OS rate was 64.7%.[10][Level of evidence C3]
A prospective multicenter study (NCT02733458) included 52 patients with newly diagnosed stage IE/IIE ENKL who received two cycles of GELAD followed by intensity-modulated radiotherapy (50–56 Gy in 25–58 fractions) and two additional courses of GELAD.[11]
With a median follow-up of 32 months, the estimated 4-year OS rate was 94.2% (95% CI, 83.2%–93.1%), and the 4-year PFS rate was 90.4% (95% CI, 78.4%–95.9%).[11][Level of evidence C3]
The overall response rate was 94.2%, and the complete response rate was 92.3%.
A retrospective study included 80 patients with newly diagnosed (n = 48), refractory (n = 23), or first-relapse (n = 9) ENKL. Patients received DDGP.[12]
The overall response rate was 91.3% (95% CI, 85.0%–96.3%), and the complete response rate was 60.0% (95% CI, 48.8%–71.3%). The 2-year PFS rate was 81.4% (95% CI, 76.3%–86.5%), and the 2-year OS rate was 87.1% (95% CI, 83.4%–91.4%).[12][Level of evidence C3]
A phase I/II study of 27 patients with newly diagnosed stage IE or contiguous IIE disease studied the nonasparaginase-based regimen DeVIC (dexamethasone, etoposide, ifosfamide, and carboplatin). Patients received three cycles of DeVIC at the recommended phase II dosing and concurrent radiotherapy dosed at 50 Gy.[13]
With a median follow-up of 32 months, the 2-year OS rate was 78% (95% CI, 57%–89%). The overall response rate was 81%, and the complete response rate was 77%.[13][Level of evidence C3]
The most common grade 3 or higher nonhematologic toxicity was mucositis related to radiation, which occurred in 30% of patients.
Radiation therapy is an essential component of treatment for localized ENKL, nasal type, whether given as a monotherapy or as part of combined modality therapy.
Evidence (radiation therapy with or without chemotherapy):
A retrospective review included 1,273 patients with early-stage disease. Patients were stratified into a low-risk group and high-risk group using stage, age, lactate dehydrogenase level, performance status, and primary tumor invasion.
Low-risk patients fared best with radiation therapy alone,[14] while high-risk patients fared best with a strategy of radiation therapy combined with chemotherapy.[15–17][Level of evidence C3]
A retrospective review included 303 previously untreated patients from an international consortium who received nonanthracycline chemotherapy.[18]
The OS rates were identical (72%−74% at 5 years) for patients with early-stage disease who received either concurrent chemotherapy and radiation therapy or chemotherapy followed by radiation therapy.[18][Level of evidence C3]
Higher doses of radiation therapy administered at more than 50 Gy are associated with improved outcomes according to anecdotal reports.[17] The highly aggressive course, with poor response and short survival with standard therapies, especially for patients with advanced-stage disease or extranasal presentation, has led some investigators to recommend autologous or allogeneic peripheral stem cell transplant consolidation.[19–24][Level of evidence C3]
For patients with relapsed or refractory disease, therapies targeting programmed death 1 (PD-1) and programmed death-ligand 1 (PD-L1) have demonstrated promising results.
Evidence (immunotherapy):
In a phase II trial, the anti–PD-L1 antibody avelumab was given to 21 patients with relapsed or refractory disease.[25]
The complete response rate was 24%, and the overall response rate was 38%. The responses correlated with tumor PD-L1 expression.[25][Level of evidence C3]
Treatment with pembrolizumab, an anti–PD-1 antibody, resulted in similar responses in patients with relapsed or refractory disease.[26][Level of evidence C3]
A single-arm multicenter phase II study (GEMSTONE-201 [NCT03595657]) included 80 patients with relapsed or refractory ENKL. Patients received the anti–PD-L1 monoclonal antibody sugemalimab.
The objective response rate was 44.9% (95% CI, 33.6%–56.6%), and the complete response rate was 35.9%. The 12-month duration of response rate was 82.5% (95% CI, 62.0%–92.6%).[27][Level of evidence B3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Tse E, Kwong YL: How I treat NK/T-cell lymphomas. Blood 121 (25): 4997-5005, 2013. [PUBMED Abstract]
Tang YT, Wang D, Luo H, et al.: Aggressive NK-cell leukemia: clinical subtypes, molecular features, and treatment outcomes. Blood Cancer J 7 (12): 660, 2017. [PUBMED Abstract]
Suzuki R, Suzumiya J, Yamaguchi M, et al.: Prognostic factors for mature natural killer (NK) cell neoplasms: aggressive NK cell leukemia and extranodal NK cell lymphoma, nasal type. Ann Oncol 21 (5): 1032-40, 2010. [PUBMED Abstract]
Qi SN, Li YX, Specht L, et al.: Modern Radiation Therapy for Extranodal Nasal-Type NK/T-cell Lymphoma: Risk-Adapted Therapy, Target Volume, and Dose Guidelines from the International Lymphoma Radiation Oncology Group. Int J Radiat Oncol Biol Phys 110 (4): 1064-1081, 2021. [PUBMED Abstract]
Obama K, Tara M, Niina K: L-asparaginase-Based induction therapy for advanced extranodal NK/T-cell lymphoma. Int J Hematol 78 (3): 248-50, 2003. [PUBMED Abstract]
Ando M, Sugimoto K, Kitoh T, et al.: Selective apoptosis of natural killer-cell tumours by l-asparaginase. Br J Haematol 130 (6): 860-8, 2005. [PUBMED Abstract]
Yamaguchi M, Kwong YL, Kim WS, et al.: Phase II study of SMILE chemotherapy for newly diagnosed stage IV, relapsed, or refractory extranodal natural killer (NK)/T-cell lymphoma, nasal type: the NK-Cell Tumor Study Group study. J Clin Oncol 29 (33): 4410-6, 2011. [PUBMED Abstract]
Ghione P, Qi S, Imber BS, et al.: Modified SMILE (mSMILE) and intensity-modulated radiotherapy (IMRT) for extranodal NK-T lymphoma nasal type in a single-center population. Leuk Lymphoma 61 (14): 3331-3341, 2020. [PUBMED Abstract]
Wang JH, Wang L, Liu CC, et al.: Efficacy of combined gemcitabine, oxaliplatin and pegaspargase (P-gemox regimen) in patients with newly diagnosed advanced-stage or relapsed/refractory extranodal NK/T-cell lymphoma. Oncotarget 7 (20): 29092-101, 2016. [PUBMED Abstract]
Zhu Y, Tian S, Xu L, et al.: GELAD chemotherapy with sandwiched radiotherapy for patients with newly diagnosed stage IE/IIE natural killer/T-cell lymphoma: a prospective multicentre study. Br J Haematol 196 (4): 939-946, 2022. [PUBMED Abstract]
Zhang L, Li S, Jia S, et al.: The DDGP (cisplatin, dexamethasone, gemcitabine, and pegaspargase) regimen for treatment of extranodal natural killer (NK)/T-cell lymphoma, nasal type. Oncotarget 7 (36): 58396-58404, 2016. [PUBMED Abstract]
Yamaguchi M, Tobinai K, Oguchi M, et al.: Phase I/II study of concurrent chemoradiotherapy for localized nasal natural killer/T-cell lymphoma: Japan Clinical Oncology Group Study JCOG0211. J Clin Oncol 27 (33): 5594-600, 2009. [PUBMED Abstract]
Yang Y, Cao JZ, Lan SM, et al.: Association of Improved Locoregional Control With Prolonged Survival in Early-Stage Extranodal Nasal-Type Natural Killer/T-Cell Lymphoma. JAMA Oncol 3 (1): 83-91, 2017. [PUBMED Abstract]
Yang Y, Zhu Y, Cao JZ, et al.: Risk-adapted therapy for early-stage extranodal nasal-type NK/T-cell lymphoma: analysis from a multicenter study. Blood 126 (12): 1424-32; quiz 1517, 2015. [PUBMED Abstract]
Yamaguchi M, Suzuki R, Oguchi M, et al.: Treatments and Outcomes of Patients With Extranodal Natural Killer/T-Cell Lymphoma Diagnosed Between 2000 and 2013: A Cooperative Study in Japan. J Clin Oncol 35 (1): 32-39, 2017. [PUBMED Abstract]
Vargo JA, Patel A, Glaser SM, et al.: The impact of the omission or inadequate dosing of radiotherapy in extranodal natural killer T-cell lymphoma, nasal type, in the United States. Cancer 123 (16): 3176-3185, 2017. [PUBMED Abstract]
Kwong YL, Kim SJ, Tse E, et al.: Sequential chemotherapy/radiotherapy was comparable with concurrent chemoradiotherapy for stage I/II NK/T-cell lymphoma. Ann Oncol 29 (1): 256-263, 2018. [PUBMED Abstract]
Liang R, Todd D, Chan TK, et al.: Treatment outcome and prognostic factors for primary nasal lymphoma. J Clin Oncol 13 (3): 666-70, 1995. [PUBMED Abstract]
Cheung MM, Chan JK, Lau WH, et al.: Primary non-Hodgkin’s lymphoma of the nose and nasopharynx: clinical features, tumor immunophenotype, and treatment outcome in 113 patients. J Clin Oncol 16 (1): 70-7, 1998. [PUBMED Abstract]
Hausdorff J, Davis E, Long G, et al.: Non-Hodgkin’s lymphoma of the paranasal sinuses: clinical and pathological features, and response to combined-modality therapy. Cancer J Sci Am 3 (5): 303-11, 1997 Sep-Oct. [PUBMED Abstract]
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008. [PUBMED Abstract]
Au WY, Weisenburger DD, Intragumtornchai T, et al.: Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood 113 (17): 3931-7, 2009. [PUBMED Abstract]
Yamaguchi M, Suzuki R, Oguchi M: Advances in the treatment of extranodal NK/T-cell lymphoma, nasal type. Blood 131 (23): 2528-2540, 2018. [PUBMED Abstract]
Kim SJ, Lim JQ, Laurensia Y, et al.: Avelumab for the treatment of relapsed or refractory extranodal NK/T-cell lymphoma: an open-label phase 2 study. Blood 136 (24): 2754-2763, 2020. [PUBMED Abstract]
Kwong YL, Chan TSY, Tan D, et al.: PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 129 (17): 2437-2442, 2017. [PUBMED Abstract]
Huang H, Tao R, Hao S, et al.: Sugemalimab Monotherapy for Patients With Relapsed or Refractory Extranodal Natural Killer/T-Cell Lymphoma (GEMSTONE-201): Results From a Single-Arm, Multicenter, Phase II Study. J Clin Oncol 41 (16): 3032-3041, 2023. [PUBMED Abstract]
Treatment of Enteropathy-Associated and Monomorphic Epitheliotropic Intestinal T-Cell Lymphomas
Enteropathy-associated T-cell lymphoma (EATL) and monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL) are rare subsets of peripheral T-cell lymphoma (PTCL) which together account for less than 5% of PTCL. EATL (sometimes referred to as type 1 EATL) is a complication of celiac disease and is more common in Europe. MEITL (sometimes referred to as type 2 EATL) is not associated with celiac disease and is more common in Asia. SETD2 pathogenic variants and, to a lesser extent, STAT5B pathogenic variants are seen in most cases of EATL and MEITL.[1,2] Because a gluten-free diet prevents the development of lymphoma in patients with celiac disease, patients diagnosed in childhood rarely develop EATL. The diagnosis of celiac disease is usually made by finding villous atrophy in the resected intestine. Surgery is often required for diagnosis and to avoid perforation during therapy.[3]
Treatment Options for Enteropathy-Associated and Monomorphic Epitheliotropic Intestinal T-Cell Lymphomas
Recommended management includes CHO(E)P (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone), but relapse is common and outcomes are generally poor.[4–6] Complications of treatment include gastrointestinal bleeding, small bowel perforation, and enterocolic fistulae. Patients often require parenteral nutrition. For more information on parenteral nutrition, see Nutrition in Cancer Care. Multifocal intestinal perforations and visceral abdominal involvement are seen at the time of relapse. Consolidation with either high-dose chemotherapy with autologous stem cell rescue or allogeneic stem cell transplant is often used in first or later remission.[4,7,8][Level of evidence C3] Evidence for this approach is anecdotal.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Veloza L, Cavalieri D, Missiaglia E, et al.: Monomorphic epitheliotropic intestinal T-cell lymphoma comprises morphologic and genomic heterogeneity impacting outcome. Haematologica 108 (1): 181-195, 2023. [PUBMED Abstract]
Moffitt AB, Ondrejka SL, McKinney M, et al.: Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J Exp Med 214 (5): 1371-1386, 2017. [PUBMED Abstract]
Egan LJ, Walsh SV, Stevens FM, et al.: Celiac-associated lymphoma. A single institution experience of 30 cases in the combination chemotherapy era. J Clin Gastroenterol 21 (2): 123-9, 1995. [PUBMED Abstract]
Gale J, Simmonds PD, Mead GM, et al.: Enteropathy-type intestinal T-cell lymphoma: clinical features and treatment of 31 patients in a single center. J Clin Oncol 18 (4): 795-803, 2000. [PUBMED Abstract]
Daum S, Ullrich R, Heise W, et al.: Intestinal non-Hodgkin’s lymphoma: a multicenter prospective clinical study from the German Study Group on Intestinal non-Hodgkin’s Lymphoma. J Clin Oncol 21 (14): 2740-6, 2003. [PUBMED Abstract]
Di Sabatino A, Biagi F, Gobbi PG, et al.: How I treat enteropathy-associated T-cell lymphoma. Blood 119 (11): 2458-68, 2012. [PUBMED Abstract]
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008. [PUBMED Abstract]
Sieniawski M, Angamuthu N, Boyd K, et al.: Evaluation of enteropathy-associated T-cell lymphoma comparing standard therapies with a novel regimen including autologous stem cell transplantation. Blood 115 (18): 3664-70, 2010. [PUBMED Abstract]
Treatment of Indolent T-Cell Lymphoma of the Gastrointestinal Tract
In contrast to the more aggressive forms of T-cell lymphoma with intestinal involvement, indolent T-cell lymphoma of the gastrointestinal tract (iTCL-GI) often presents more indolently. iTCL-GI can be difficult to distinguish from inflammatory bowel disease or other autoimmune disorders with gastrointestinal involvement.[1,2]
iTCL-GI is often diagnosed when multiple biopsies from different sites show matching clones by T-cell receptor gene rearrangement testing. While the immunophenotype can vary (iTCL-GI is frequently CD4+ but can also be CD8+ or CD4-/CD8-), Ki-67 is typically very low at 5% to 10%. Unlike some other intestinal lymphomas, Epstein-Barr encoding region staining for Epstein-Barr virus is usually negative. Although initial reports of this rare entity suggested a low likelihood of transformation to a more aggressive process, transformation to peripheral T-cell lymphoma, not otherwise specific and other aggressive forms has since been documented.[3]
Treatment Options for Indolent T-Cell Lymphoma of the Gastrointestinal Tract
Response to multiagent chemotherapy is limited, and many patients can be managed with watchful waiting alone. Limited studies suggest that alterations in the JAK/STAT pathway or genes involved in epigenetic modification could serve as potential therapeutic targets.[4]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Margolskee E, Jobanputra V, Lewis SK, et al.: Indolent small intestinal CD4+ T-cell lymphoma is a distinct entity with unique biologic and clinical features. PLoS One 8 (7): e68343, 2013. [PUBMED Abstract]
Perry AM, Warnke RA, Hu Q, et al.: Indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Blood 122 (22): 3599-606, 2013. [PUBMED Abstract]
Sanguedolce F, Zanelli M, Zizzo M, et al.: Indolent T-Cell Lymphoproliferative Disorders of the Gastrointestinal Tract (iTLPD-GI): A Review. Cancers (Basel) 13 (11): , 2021. [PUBMED Abstract]
Sharma A, Oishi N, Boddicker RL, et al.: Recurrent STAT3-JAK2 fusions in indolent T-cell lymphoproliferative disorder of the gastrointestinal tract. Blood 131 (20): 2262-2266, 2018. [PUBMED Abstract]
Treatment of Hepatosplenic T-Cell Lymphoma
Hepatosplenic T-cell lymphoma (HSTCL) is a rare subtype of peripheral T-cell lymphoma (PTCL) comprising approximately 1% of PTCL. HSTCL often involves young men. HSTCL appears to be localized to the hepatic and splenic sinusoids, with cell surface expression of the gamma delta T-cell receptor.[1–3] Characteristic chromosomal abnormalities such as isochromosome 7q and trisomy 8 are also suggestive of the diagnosis.[4] While in most cases the neoplastic cells express a gamma delta T-cell receptor (hepatosplenic gamma delta T-cell lymphoma), there are reports of alpha beta T-cell receptor expression (hepatosplenic alpha beta T-cell lymphoma).[5] This lymphoma has an extremely poor prognosis and an extremely aggressive clinical course.
Treatment Options for Hepatosplenic T-Cell Lymphoma
HSTCL is treated with induction chemotherapy and stem cell transplant (SCT) consolidation.[3,6]
Evidence (induction chemotherapy and SCT consolidation):
A meta-analysis of 118 patients with HSTCL compared CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) and CHOP-like induction regimens.[7]
Non–CHOP-based regimens (containing cytarabine, etoposide, and/or platinum-based treatment) were associated with improved outcomes, including an overall response rate of 82% versus 52% (P = .006) and a median overall survival (OS) of 36.5 months versus 18 months (P = .00014).
Consolidation with allogeneic SCT was associated with an improved median OS of 33 months, versus 27 months (P = .016) for autologous SCT.[7][Level of evidence C3]
The use of ICE (ifosfamide, carboplatin, and etoposide) or IVAC (ifosfamide, etoposide, and high-dose cytarabine) has resulted in improved responses when compared with CHOP in other smaller studies as well.[8][Level of evidence D] Given the inadequate responses to CHOP or CHOP-like regimens, many clinicians are using ICE chemotherapy as front-line induction therapy, followed by consolidative allogeneic SCT for patients achieving a first remission. However, the efficacy of this approach is undetermined.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Belhadj K, Reyes F, Farcet JP, et al.: Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood 102 (13): 4261-9, 2003. [PUBMED Abstract]
Chanan-Khan A, Islam T, Alam A, et al.: Long-term survival with allogeneic stem cell transplant and donor lymphocyte infusion following salvage therapy with anti-CD52 monoclonal antibody (Campath) in a patient with alpha/beta hepatosplenic T-cell non-Hodgkin’s lymphoma. Leuk Lymphoma 45 (8): 1673-5, 2004. [PUBMED Abstract]
Pro B, Allen P, Behdad A: Hepatosplenic T-cell lymphoma: a rare but challenging entity. Blood 136 (18): 2018-2026, 2020. [PUBMED Abstract]
Travert M, Huang Y, de Leval L, et al.: Molecular features of hepatosplenic T-cell lymphoma unravels potential novel therapeutic targets. Blood 119 (24): 5795-806, 2012. [PUBMED Abstract]
Macon WR, Levy NB, Kurtin PJ, et al.: Hepatosplenic alphabeta T-cell lymphomas: a report of 14 cases and comparison with hepatosplenic gammadelta T-cell lymphomas. Am J Surg Pathol 25 (3): 285-96, 2001. [PUBMED Abstract]
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008. [PUBMED Abstract]
Klebaner D, Koura D, Tzachanis D, et al.: Intensive Induction Therapy Compared With CHOP for Hepatosplenic T-cell Lymphoma. Clin Lymphoma Myeloma Leuk 20 (7): 431-437.e2, 2020. [PUBMED Abstract]
Voss MH, Lunning MA, Maragulia JC, et al.: Intensive induction chemotherapy followed by early high-dose therapy and hematopoietic stem cell transplantation results in improved outcome for patients with hepatosplenic T-cell lymphoma: a single institution experience. Clin Lymphoma Myeloma Leuk 13 (1): 8-14, 2013. [PUBMED Abstract]
Treatment of Adult T-Cell Leukemia/Lymphoma
Adult T-cell leukemia/lymphoma (ATL) is caused by infection with the retrovirus human T-lymphotrophic virus 1 (HTLV1) and is frequently associated with lymphadenopathy, hypercalcemia, circulating leukemic cells, bone and skin involvement, hepatosplenomegaly, a rapidly progressive course, and poor response to combination chemotherapy.[1,2] HTLV1 is endemic to Japan (especially the southern island of Kyushu), central Africa, the Caribbean islands, and some regions of Central and South America, the Middle East, and Australia. This retrovirus is mostly spread by sexual contact or breastfeeding.[3] ATL has been divided into four clinical subtypes:[3]
Acute (aggressive course with leukemia, with or without extranodal or nodal involvement).
Lymphoma (aggressive course with lymphadenopathy and no leukemia).
Chronic (indolent course with leukemia and lymphadenopathy).
Smoldering (indolent course with only leukemia).
Treatment Options for Adult T-Cell Leukemia/Lymphoma
The anti-CCR4 monoclonal antibody mogamulizumab has a targeted lysis of the ATL clone and also reduces the immunosuppressive Treg population, allowing improvement of cell-mediated immunotherapy.[4] Single-agent phase II trials in patients with ATL showed response rates ranging from 30% to 50%.[5–7][Level of evidence C3] In a phase II study that combined mogamulizumab with combination chemotherapy, 101 patients had an overall response rate of 65% (95% confidence interval [CI], 55%–75%), a median progression-free survival of 7.4 months (95% CI, 5.7–9.1), and a median overall survival (OS) of 16 months (95% CI, 10.3–21.8).[8][Level of evidence C3]
A retrospective review evaluated mogamulizumab combined with both the VCAP-AMP-VECP (vincristine, cyclophosphamide, doxorubicin, prednisone; doxorubicin, ranimustine, prednisone; vindesine, etoposide, carboplatin, prednisone) regimen and CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone)-based regimens versus chemotherapy alone. The review concluded that the 4-year OS rate was 46.3% in the mogamulizumab combination groups, compared with 20.6% in the chemotherapy-alone group.[9][Level of evidence C3] The best results were seen with CHOP-based regimens. Mogamulizumab is available in the United States but has not been approved by the U.S. Food and Drug Administration (FDA) for patients with ATL.
Mogamulizumab is often avoided in patients planning allogeneic SCT based on data from Japan that showed an increased risk of severe graft-versus-host disease (GVHD) in patients who received mogamulizumab before allogeneic SCT.[10] The relevance of these findings in other countries and the impact of different GVHD prophylaxis regimens in this setting remains to be determined.
Brentuximab vedotin
Brentuximab vedotin is a monoclonal antibody-drug conjugate directed at CD30 and is a treatment option for patients with ATL whose tumor expresses CD30.[11] Patients with ATL were included in a clinical trial of brentuximab vedotin that mostly involved patients with anaplastic large T-cell lymphoma. A statistical benefit for the very small ATL subgroup could not be established.[11][Level of evidence C3]
Lenalidomide
In a multicenter phase II study (NCT01724177) of 26 patients with relapsed ATL, the overall response rate was 42% (95% CI, 23%–63%) in patients who received the immunomodulatory agent lenalidomide.[12][Level of evidence C3]
Valemetostat
Valemetostat is a selective dual inhibitor of EZH1 and EZH2, which are important histone methyltransferases involved with chromatin folding.[13] A multicenter phase II study (NCT04102150) enrolled 25 patients with relapsed or refractory ATL, 24 of whom had prior therapy with mogamulizumab.[14] With a median follow-up of 6.5 months, the overall response rate was 48% (90% CI, 30.5%–65.9%). There were five complete remissions and seven partial remissions among the heavily pretreated patients.[14][Level of evidence C3] Responses were seen in patients with the acute, lymphoma, and chronic versions of ATL. Although approved for use in Japan, valemetostat is not approved by the FDA for use in the United States.
Zidovudine and interferon alfa
The combination of zidovudine and interferon alfa has activity against ATL, especially among patients with the chronic and smoldering versions of ATL.[15,16] Durable remissions of 8 to 12 months and response rates of 20% to 40% have been reported for patients, some of whom had relapsed disease after prior chemotherapy,[15,16] and some with the acute and lymphoma subtypes of ATL.[17,18]
Cytotoxic chemotherapy and allogeneic SCT
The acute and lymphoma types of ATL respond poorly to combination cytotoxic chemotherapy and allogeneic SCT, with a median OS under 1 year.[19–21] Less than 10% of patients who received combination therapy were alive after 4 years in one retrospective study.[21] Durable remissions have been reported after allogeneic SCT and even after subsequent donor lymphocyte infusion for relapses after transplant.[22–24][Level of evidence C3] Among 815 patients who underwent allogeneic SCT in two retrospective reviews, the 3-year OS rates were 36% and 26%.[25,26][Level of evidence C1]
The cytotoxic chemotherapy backbone for patients with ATL in the United States and Europe is the CHOP or CHO(E)P regimen (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone). Brentuximab vedotin has been added to CHOP in rare cases when patients express CD30. For the acute and lymphoma versions of ATL, a series of clinical trials in Japan established VCAP-AMP-VECP as the standard first-line treatment.[2,27,28] This regimen has generally not been implemented outside of Asia since a trial comparing VCAP-AMP-VECP versus CHOP-14 did not show statistically improved OS (3-year OS rate, 24% vs. 13%; P = nonsignificant).
Allogeneic SCT is frequently used for patients achieving first or later remission, although results are largely anecdotal.[29,30]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Bangham CRM: HTLV-1 persistence and the oncogenesis of adult T-cell leukemia/lymphoma. Blood 141 (19): 2299-2306, 2023. [PUBMED Abstract]
Cook LB, Phillips AA: How I treat adult T-cell leukemia/lymphoma. Blood 137 (4): 459-470, 2021. [PUBMED Abstract]
Horwitz S, O’Connor OA, Pro B, et al.: Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393 (10168): 229-240, 2019. [PUBMED Abstract]
Ishida T, Fujiwara H, Nosaka K, et al.: Multicenter Phase II Study of Lenalidomide in Relapsed or Recurrent Adult T-Cell Leukemia/Lymphoma: ATLL-002. J Clin Oncol 34 (34): 4086-4093, 2016. [PUBMED Abstract]
Izutsu K, Makita S, Nosaka K, et al.: An open-label, single-arm phase 2 trial of valemetostat for relapsed or refractory adult T-cell leukemia/lymphoma. Blood 141 (10): 1159-1168, 2023. [PUBMED Abstract]
Bazarbachi A, Plumelle Y, Carlos Ramos J, et al.: Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J Clin Oncol 28 (27): 4177-83, 2010. [PUBMED Abstract]
Bazarbachi A, Suarez F, Fields P, et al.: How I treat adult T-cell leukemia/lymphoma. Blood 118 (7): 1736-45, 2011. [PUBMED Abstract]
Cook LB, Rowan AG, Demontis MA, et al.: Long-term clinical remission maintained after cessation of zidovudine and interferon-α therapy in chronic adult T-cell leukemia/lymphoma. Int J Hematol 107 (3): 378-382, 2018. [PUBMED Abstract]
Hodson A, Crichton S, Montoto S, et al.: Use of zidovudine and interferon alfa with chemotherapy improves survival in both acute and lymphoma subtypes of adult T-cell leukemia/lymphoma. J Clin Oncol 29 (35): 4696-701, 2011. [PUBMED Abstract]
Yamada Y, Tomonaga M, Fukuda H, et al.: A new G-CSF-supported combination chemotherapy, LSG15, for adult T-cell leukaemia-lymphoma: Japan Clinical Oncology Group Study 9303. Br J Haematol 113 (2): 375-82, 2001. [PUBMED Abstract]
Fukushima T, Miyazaki Y, Honda S, et al.: Allogeneic hematopoietic stem cell transplantation provides sustained long-term survival for patients with adult T-cell leukemia/lymphoma. Leukemia 19 (5): 829-34, 2005. [PUBMED Abstract]
Katsuya H, Yamanaka T, Ishitsuka K, et al.: Prognostic index for acute- and lymphoma-type adult T-cell leukemia/lymphoma. J Clin Oncol 30 (14): 1635-40, 2012. [PUBMED Abstract]
Itonaga H, Tsushima H, Taguchi J, et al.: Treatment of relapsed adult T-cell leukemia/lymphoma after allogeneic hematopoietic stem cell transplantation: the Nagasaki Transplant Group experience. Blood 121 (1): 219-25, 2013. [PUBMED Abstract]
Cook LB, Fuji S, Hermine O, et al.: Revised Adult T-Cell Leukemia-Lymphoma International Consensus Meeting Report. J Clin Oncol 37 (8): 677-687, 2019. [PUBMED Abstract]
Yoshimitsu M, Tanosaki R, Kato K, et al.: Risk Assessment in Adult T Cell Leukemia/Lymphoma Treated with Allogeneic Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant 24 (4): 832-839, 2018. [PUBMED Abstract]
Ishida T, Hishizawa M, Kato K, et al.: Allogeneic hematopoietic stem cell transplantation for adult T-cell leukemia-lymphoma with special emphasis on preconditioning regimen: a nationwide retrospective study. Blood 120 (8): 1734-41, 2012. [PUBMED Abstract]
Katsuya H, Ishitsuka K, Utsunomiya A, et al.: Treatment and survival among 1594 patients with ATL. Blood 126 (24): 2570-7, 2015. [PUBMED Abstract]
Hermine O, Ramos JC, Tobinai K: A Review of New Findings in Adult T-cell Leukemia-Lymphoma: A Focus on Current and Emerging Treatment Strategies. Adv Ther 35 (2): 135-152, 2018. [PUBMED Abstract]
Tsukasaki K, Utsunomiya A, Fukuda H, et al.: VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study JCOG9801. J Clin Oncol 25 (34): 5458-64, 2007. [PUBMED Abstract]
Bazarbachi AH, Reef D, Narvel H, et al.: Outcome of Stem Cell Transplantation in HTLV-1-Associated North American Adult T-Cell Leukemia/Lymphoma. Clin Hematol Int 5 (2-3): 78-91, 2023. [PUBMED Abstract]
Tanase AD, Colita A, Craciun OG, et al.: Allogeneic Stem Cell Transplantation for Adult T-Cell Leukemia/Lymphoma-Romanian Experience. J Clin Med 9 (8): , 2020. [PUBMED Abstract]
Treatment of T-Cell Prolymphocytic Leukemia
Prolymphocytic leukemia (PLL) is a rare form of lymphocytic leukemia characterized by excessive prolymphocytes in the blood with a typical phenotype that is positive for CD19, CD20, and surface-membrane immunoglobulin and negative for CD5.[1] These patients demonstrate splenomegaly and poor response to low-dose or high-dose chemotherapy.[2,3]
Treatment Options for T-Cell Prolymphocytic Leukemia
Cladribine appears to be an active agent (60% complete remission rate) for patients with de novo B-cell PLL.[4][Level of evidence C3] Anecdotal responses have been seen with venetoclax.[5][Level of evidence C3] Alemtuzumab, an anti-CD52 humanized monoclonal antibody, has been used for 76 patients with T-cell PLL after failure of previous chemotherapy (usually pentostatin or cladribine). The response rate was 51% (95% confidence interval, 40%–63%), and the median time to progression was 4.5 months (range, 0.1–45.4).[6][Level of evidence C3] These response rates have been confirmed by other investigators.[7] Patients with chronic lymphocytic leukemia (CLL) and prolymphocytoid transformation maintain the classic CLL phenotype and have a worse prognosis than patients with PLL.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Staber PB, Herling M, Bellido M, et al.: Consensus criteria for diagnosis, staging, and treatment response assessment of T-cell prolymphocytic leukemia. Blood 134 (14): 1132-1143, 2019. [PUBMED Abstract]
Rozman C, Montserrat E: Chronic lymphocytic leukemia. N Engl J Med 333 (16): 1052-7, 1995. [PUBMED Abstract]
Melo JV, Catovsky D, Galton DA: The relationship between chronic lymphocytic leukaemia and prolymphocytic leukaemia. I. Clinical and laboratory features of 300 patients and characterization of an intermediate group. Br J Haematol 63 (2): 377-87, 1986. [PUBMED Abstract]
Saven A, Lee T, Schlutz M, et al.: Major activity of cladribine in patients with de novo B-cell prolymphocytic leukemia. J Clin Oncol 15 (1): 37-43, 1997. [PUBMED Abstract]
Boidol B, Kornauth C, van der Kouwe E, et al.: First-in-human response of BCL-2 inhibitor venetoclax in T-cell prolymphocytic leukemia. Blood 130 (23): 2499-2503, 2017. [PUBMED Abstract]
Keating MJ, Cazin B, Coutré S, et al.: Campath-1H treatment of T-cell prolymphocytic leukemia in patients for whom at least one prior chemotherapy regimen has failed. J Clin Oncol 20 (1): 205-13, 2002. [PUBMED Abstract]
Dearden CE, Matutes E, Catovsky D: Alemtuzumab in T-cell malignancies. Med Oncol 19 (Suppl): S27-32, 2002. [PUBMED Abstract]
Treatment of Relapsed or Refractory Peripheral T-Cell Lymphoma
Treatment Options for Relapsed or Refractory Peripheral T-Cell Lymphoma
Treatment options for relapsed or refractory peripheral T-cell lymphoma include:
Combination chemotherapy.
ICE (ifosfamide, carboplatin, and etoposide).[1,2]
GEMOX (gemcitabine, oxaliplatin, and dexamethasone).[3]
DHAP (dexamethasone, high-dose cytarabine, and cisplatin).[4]
ESHAP (etoposide, methylprednisolone, high-dose cytarabine, and cisplatin).[5,6]
Hyper-CVAD (cyclophosphamide, vincristine, doxorubicin, and dexamethasone).[7]
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Jerkeman M, Leppä S, Kvaløy S, et al.: ICE (ifosfamide, carboplatin, etoposide) as second-line chemotherapy in relapsed or primary progressive aggressive lymphoma–the Nordic Lymphoma Group experience. Eur J Haematol 73 (3): 179-82, 2004. [PUBMED Abstract]
Zelenetz AD, Hamlin P, Kewalramani T, et al.: Ifosfamide, carboplatin, etoposide (ICE)-based second-line chemotherapy for the management of relapsed and refractory aggressive non-Hodgkin’s lymphoma. Ann Oncol 14 (Suppl 1): i5-10, 2003. [PUBMED Abstract]
Shen QD, Wang L, Zhu HY, et al.: Gemcitabine, oxaliplatin and dexamethasone (GemDOx) as salvage therapy for relapsed or refractory diffuse large B-cell lymphoma and peripheral T-cell lymphoma. J Cancer 12 (1): 163-169, 2021. [PUBMED Abstract]
Schmitz N, Truemper L, Bouabdallah K, et al.: A randomized phase 3 trial of autologous vs allogeneic transplantation as part of first-line therapy in poor-risk peripheral T-NHL. Blood 137 (19): 2646-2656, 2021. [PUBMED Abstract]
Velasquez WS, McLaughlin P, Tucker S, et al.: ESHAP–an effective chemotherapy regimen in refractory and relapsing lymphoma: a 4-year follow-up study. J Clin Oncol 12 (6): 1169-76, 1994. [PUBMED Abstract]
Mercadal S, Briones J, Xicoy B, et al.: Intensive chemotherapy (high-dose CHOP/ESHAP regimen) followed by autologous stem-cell transplantation in previously untreated patients with peripheral T-cell lymphoma. Ann Oncol 19 (5): 958-63, 2008. [PUBMED Abstract]
Hapgood G, Stone JM, Zannino D, et al.: A phase II study of a modified hyper-CVAD frontline therapy for patients with adverse risk diffuse large B-cell and peripheral T-cell non-Hodgkin lymphoma. Leuk Lymphoma 60 (4): 904-911, 2019. [PUBMED Abstract]
Iyer SP, Foss FF: Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. Oncologist 20 (9): 1084-91, 2015. [PUBMED Abstract]
Campbell P, Thomas CM: Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. J Oncol Pharm Pract 23 (2): 143-147, 2017. [PUBMED Abstract]
O’Connor OA, Horwitz S, Masszi T, et al.: Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 33 (23): 2492-9, 2015. [PUBMED Abstract]
O’Connor OA, Pro B, Pinter-Brown L, et al.: Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 29 (9): 1182-9, 2011. [PUBMED Abstract]
Chen AI, McMillan A, Negrin RS, et al.: Long-term results of autologous hematopoietic cell transplantation for peripheral T cell lymphoma: the Stanford experience. Biol Blood Marrow Transplant 14 (7): 741-7, 2008. [PUBMED Abstract]
Hamadani M, Ngoya M, Sureda A, et al.: Outcome of allogeneic transplantation for mature T-cell lymphomas: impact of donor source and disease characteristics. Blood Adv 6 (3): 920-930, 2022. [PUBMED Abstract]
Mehta-Shah N, Kommalapati A, Teja S: Successful treatment of mature T-cell lymphoma with allogeneic stem cell transplantation: the largest multicenter retrospective analysis. [Abstract] Blood 136 (Suppl 1): A-624, 35-36, 2020.
Sterling CH, Hughes MS, Tsai HL, et al.: Allogeneic Blood or Marrow Transplantation with Post-Transplantation Cyclophosphamide for Peripheral T Cell Lymphoma: The Importance of Graft Source. Transplant Cell Ther 29 (4): 267.e1-267.e5, 2023. [PUBMED Abstract]
Latest Updates to This Summary (05/13/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of adult peripheral T-cell non-Hodgkin lymphoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Peripheral T-Cell Non-Hodgkin Lymphoma Treatment are:
Eric J. Seifter, MD (Johns Hopkins University)
Cole H. Sterling, MD (Johns Hopkins University)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Peripheral T-Cell Non-Hodgkin Lymphoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/lymphoma/hp/peripheral-t-cell-lymphoma-pdq. Accessed <MM/DD/YYYY>. [PMID: 37437079]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Like Hodgkin lymphoma, NHL usually originates in lymphoid tissues and can spread to other organs. However, NHL is much less predictable than Hodgkin lymphoma and has a far greater tendency to spread to extranodal sites. The prognosis depends on the histological type, disease stage, and treatment.
Incidence and Mortality
Estimated new cases and deaths from all types of NHL in the United States in 2025:[2]
New cases: 80,350.
Deaths: 19,390.
B-cell lymphomas make up about 85% of NHL cases.[3]
Anatomy
NHL usually originates in lymphoid tissues.
EnlargeThe lymph system is part of the body’s immune system and is made up of tissues and organs that help protect the body from infection and disease. These include the tonsils, adenoids (not shown), thymus, spleen, bone marrow, lymph vessels, and lymph nodes. Lymph tissue is also found in many other parts of the body, including the small intestine.
Prognosis and Survival
NHL can be divided into two prognostic groups: indolent lymphomas and aggressive lymphomas.
Indolent NHL has a relatively good prognosis, with a median survival as long as 20 years, but it is usually not curable in advanced clinical stages.[4] Early-stage (stage I and stage II) indolent NHL can be effectively treated with radiation therapy alone. Most of the indolent NHLs are nodular (or follicular) in morphology.
Aggressive NHL has a worse prognosis in the short term, but a significant number of patients can be cured with intensive combination chemotherapy regimens.
More than 70% of patients with aggressive NHL can be cured. Most relapses occur in the first 2 years after therapy. The risk of late relapse is higher in patients who manifest both indolent and aggressive histologies.[5]
Although indolent NHL responds quickly to immunotherapy, radiation therapy, and chemotherapy, a continuous rate of relapse is usually seen in advanced stages. However, patients can often be re-treated with considerable success if the disease histology remains low grade. Patients who present with, or convert to, aggressive forms of NHL may have sustained complete remissions with combination chemotherapy regimens or aggressive consolidation with marrow or stem cell support.[6,7]
Late Effects of Treatment of NHL
Late effects of treatment of non-Hodgkin lymphoma (NHL) have been observed. Impaired fertility may occur after exposure to alkylating agents.[8] For as many as three decades after diagnosis, patients are at a significantly elevated risk of developing second primary cancers, especially the following:[9–12]
Lung cancer.
Brain cancer.
Kidney cancer.
Bladder cancer.
Melanoma.
Hodgkin lymphoma.
Acute nonlymphocytic leukemia.
Left ventricular dysfunction was a significant late effect in long-term survivors of high-grade NHL who received more than 200 mg/m² of doxorubicin.[8,13]
Myelodysplastic syndrome and acute myelogenous leukemia are late complications of myeloablative therapy with autologous bone marrow or peripheral blood stem cell support, as well as conventional chemotherapy-containing alkylating agents.[10,14–21] Most of these patients show clonal hematopoiesis even before the transplant, suggesting that the hematologic injury usually occurs during induction or reinduction chemotherapy.[16,22,23] A series of 605 patients who received autologous bone marrow transplant (BMT) with cyclophosphamide and total-body radiation therapy (as conditioning) were followed for a median of 10 years. The incidence of a second malignancy was 21%, and 10% of those malignancies were solid tumors.[24]
A study of young women who received autologous BMT reported successful pregnancies with children born free of congenital abnormalities.[25] Late-occurring venous thromboembolism can occur after allogeneic or autologous BMT.[26]
Some patients have osteopenia or osteoporosis at the start of therapy; bone density may worsen after therapy for lymphoma.[27]
Long-term impaired immune health was evaluated in a retrospective cohort study of 21,690 survivors of diffuse large B-cell lymphoma from the California Cancer Registry. Elevated incidence rate ratios were found up to 10 years later for pneumonia (10.8-fold), meningitis (5.3-fold), immunoglobulin deficiency (17.6-fold), and autoimmune cytopenias (12-fold).[28] Similarly, there are impaired humoral responses to COVID-19 virus vaccination in patients with lymphoma who receive B-cell–directed therapies.[29,30]
American Cancer Society: Cancer Facts and Figures 2025. American Cancer Society, 2025. Available online. Last accessed January 16, 2025.
American Cancer Society: Types of B-cell Lymphoma. American Cancer Society, 2019. Available online. Last accessed February 7, 2025.
Tan D, Horning SJ, Hoppe RT, et al.: Improvements in observed and relative survival in follicular grade 1-2 lymphoma during 4 decades: the Stanford University experience. Blood 122 (6): 981-7, 2013. [PUBMED Abstract]
Cabanillas F, Velasquez WS, Hagemeister FB, et al.: Clinical, biologic, and histologic features of late relapses in diffuse large cell lymphoma. Blood 79 (4): 1024-8, 1992. [PUBMED Abstract]
Bastion Y, Sebban C, Berger F, et al.: Incidence, predictive factors, and outcome of lymphoma transformation in follicular lymphoma patients. J Clin Oncol 15 (4): 1587-94, 1997. [PUBMED Abstract]
Yuen AR, Kamel OW, Halpern J, et al.: Long-term survival after histologic transformation of low-grade follicular lymphoma. J Clin Oncol 13 (7): 1726-33, 1995. [PUBMED Abstract]
Haddy TB, Adde MA, McCalla J, et al.: Late effects in long-term survivors of high-grade non-Hodgkin’s lymphomas. J Clin Oncol 16 (6): 2070-9, 1998. [PUBMED Abstract]
Travis LB, Curtis RE, Glimelius B, et al.: Second cancers among long-term survivors of non-Hodgkin’s lymphoma. J Natl Cancer Inst 85 (23): 1932-7, 1993. [PUBMED Abstract]
Mudie NY, Swerdlow AJ, Higgins CD, et al.: Risk of second malignancy after non-Hodgkin’s lymphoma: a British Cohort Study. J Clin Oncol 24 (10): 1568-74, 2006. [PUBMED Abstract]
Hemminki K, Lenner P, Sundquist J, et al.: Risk of subsequent solid tumors after non-Hodgkin’s lymphoma: effect of diagnostic age and time since diagnosis. J Clin Oncol 26 (11): 1850-7, 2008. [PUBMED Abstract]
Major A, Smith DE, Ghosh D, et al.: Risk and subtypes of secondary primary malignancies in diffuse large B-cell lymphoma survivors change over time based on stage at diagnosis. Cancer 126 (1): 189-201, 2020. [PUBMED Abstract]
Moser EC, Noordijk EM, van Leeuwen FE, et al.: Long-term risk of cardiovascular disease after treatment for aggressive non-Hodgkin lymphoma. Blood 107 (7): 2912-9, 2006. [PUBMED Abstract]
Darrington DL, Vose JM, Anderson JR, et al.: Incidence and characterization of secondary myelodysplastic syndrome and acute myelogenous leukemia following high-dose chemoradiotherapy and autologous stem-cell transplantation for lymphoid malignancies. J Clin Oncol 12 (12): 2527-34, 1994. [PUBMED Abstract]
Stone RM, Neuberg D, Soiffer R, et al.: Myelodysplastic syndrome as a late complication following autologous bone marrow transplantation for non-Hodgkin’s lymphoma. J Clin Oncol 12 (12): 2535-42, 1994. [PUBMED Abstract]
Armitage JO, Carbone PP, Connors JM, et al.: Treatment-related myelodysplasia and acute leukemia in non-Hodgkin’s lymphoma patients. J Clin Oncol 21 (5): 897-906, 2003. [PUBMED Abstract]
André M, Mounier N, Leleu X, et al.: Second cancers and late toxicities after treatment of aggressive non-Hodgkin lymphoma with the ACVBP regimen: a GELA cohort study on 2837 patients. Blood 103 (4): 1222-8, 2004. [PUBMED Abstract]
Oddou S, Vey N, Viens P, et al.: Second neoplasms following high-dose chemotherapy and autologous stem cell transplantation for malignant lymphomas: a report of six cases in a cohort of 171 patients from a single institution. Leuk Lymphoma 31 (1-2): 187-94, 1998. [PUBMED Abstract]
Lenz G, Dreyling M, Schiegnitz E, et al.: Moderate increase of secondary hematologic malignancies after myeloablative radiochemotherapy and autologous stem-cell transplantation in patients with indolent lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group. J Clin Oncol 22 (24): 4926-33, 2004. [PUBMED Abstract]
McLaughlin P, Estey E, Glassman A, et al.: Myelodysplasia and acute myeloid leukemia following therapy for indolent lymphoma with fludarabine, mitoxantrone, and dexamethasone (FND) plus rituximab and interferon alpha. Blood 105 (12): 4573-5, 2005. [PUBMED Abstract]
Morton LM, Curtis RE, Linet MS, et al.: Second malignancy risks after non-Hodgkin’s lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 28 (33): 4935-44, 2010. [PUBMED Abstract]
Mach-Pascual S, Legare RD, Lu D, et al.: Predictive value of clonality assays in patients with non-Hodgkin’s lymphoma undergoing autologous bone marrow transplant: a single institution study. Blood 91 (12): 4496-503, 1998. [PUBMED Abstract]
Lillington DM, Micallef IN, Carpenter E, et al.: Detection of chromosome abnormalities pre-high-dose treatment in patients developing therapy-related myelodysplasia and secondary acute myelogenous leukemia after treatment for non-Hodgkin’s lymphoma. J Clin Oncol 19 (9): 2472-81, 2001. [PUBMED Abstract]
Brown JR, Yeckes H, Friedberg JW, et al.: Increasing incidence of late second malignancies after conditioning with cyclophosphamide and total-body irradiation and autologous bone marrow transplantation for non-Hodgkin’s lymphoma. J Clin Oncol 23 (10): 2208-14, 2005. [PUBMED Abstract]
Jackson GH, Wood A, Taylor PR, et al.: Early high dose chemotherapy intensification with autologous bone marrow transplantation in lymphoma associated with retention of fertility and normal pregnancies in females. Scotland and Newcastle Lymphoma Group, UK. Leuk Lymphoma 28 (1-2): 127-32, 1997. [PUBMED Abstract]
Gangaraju R, Chen Y, Hageman L, et al.: Risk of venous thromboembolism in patients with non-Hodgkin lymphoma surviving blood or marrow transplantation. Cancer 125 (24): 4498-4508, 2019. [PUBMED Abstract]
Westin JR, Thompson MA, Cataldo VD, et al.: Zoledronic acid for prevention of bone loss in patients receiving primary therapy for lymphomas: a prospective, randomized controlled phase III trial. Clin Lymphoma Myeloma Leuk 13 (2): 99-105, 2013. [PUBMED Abstract]
Shree T, Li Q, Glaser SL, et al.: Impaired Immune Health in Survivors of Diffuse Large B-Cell Lymphoma. J Clin Oncol 38 (15): 1664-1675, 2020. [PUBMED Abstract]
Ghione P, Gu JJ, Attwood K, et al.: Impaired humoral responses to COVID-19 vaccination in patients with lymphoma receiving B-cell-directed therapies. Blood 138 (9): 811-814, 2021. [PUBMED Abstract]
Terpos E, Trougakos IP, Gavriatopoulou M, et al.: Low neutralizing antibody responses against SARS-CoV-2 in older patients with myeloma after the first BNT162b2 vaccine dose. Blood 137 (26): 3674-3676, 2021. [PUBMED Abstract]
Cellular Classification of B-Cell Non-Hodgkin Lymphoma
A pathologist should be consulted before a biopsy because some studies require special preparation of tissue (e.g., frozen tissue). Knowledge of cell surface markers and immunoglobulin and T-cell receptor gene rearrangements may help with diagnostic and therapeutic decisions. The clonal excess of light-chain immunoglobulin may differentiate malignant cells from reactive cells. Because the prognosis and the approach to treatment are influenced by histopathology, outside biopsy specimens should be carefully reviewed by a hematopathologist who is experienced in diagnosing lymphomas. Although lymph node biopsies are recommended whenever possible, sometimes immunophenotypic data are sufficient for diagnosis of lymphoma when fine-needle aspiration cytology or core needle biopsy is preferred.[1,2]
Historical Classification Systems
Historically, uniform treatment of patients with non-Hodgkin lymphoma (NHL) has been hampered by the lack of a uniform classification system. In 1982, results of a consensus study were published as the Working Formulation.[3] The Working Formulation combined results from six major classification systems into one classification. This allowed comparison of studies from different institutions and countries. The Rappaport classification, which also follows, is no longer in common use.
Table 1. Historical Classification Systems for Non-Hodgkin Lymphoma (NHL)
A. Small lymphocytic, consistent with chronic lymphocytic leukemia
Diffuse lymphocytic, well-differentiated
B. Follicular, predominantly small-cleaved cell
Nodular lymphocytic, poorly differentiated
C. Follicular, mixed small-cleaved, and large cell
Nodular mixed, lymphocytic, and histiocytic
Intermediate grade
D. Follicular, predominantly large cell
Nodular histiocytic
E. Diffuse small-cleaved cell
Diffuse lymphocytic, poorly differentiated
F. Diffuse mixed, small and large cell
Diffuse mixed, lymphocytic, and histiocytic
G. Diffuse, large cell, cleaved, or noncleaved cell
Diffuse histiocytic
High grade
H. Immunoblastic, large cell
Diffuse histiocytic
I. Lymphoblastic, convoluted, or nonconvoluted cell
Diffuse lymphoblastic
J. Small noncleaved-cell, Burkitt, or non-Burkitt
Diffuse undifferentiated Burkitt or non-Burkitt
Current Classification Systems
As the histopathological diagnosis of NHL has become more sophisticated with the use of immunologic and genetic techniques, a number of new pathological entities have been described.[4] In addition, the understanding and treatment of many of the previously described pathological subtypes have changed. As a result, the Working Formulation has become outdated and less useful to clinicians and pathologists. European and American pathologists have proposed a new classification, the Revised European American Lymphoma (REAL) classification.[5–8] Since 1995, members of the European and American Hematopathology societies have been collaborating on a new World Health Organization (WHO) classification, which represents an updated version of the REAL system.[9,10]
Updated REAL/WHO classification
The World Health Organization (WHO) modification of the Revised European American Lymphoma (REAL) classification recognizes three major categories of lymphoid malignancies based on morphology and cell lineage: B-cell neoplasms, T-cell/natural killer (NK)-cell neoplasms, and Hodgkin lymphoma (HL). Both lymphomas and lymphoid leukemias are included in this classification because both solid and circulating phases are present in many lymphoid neoplasms and distinction between them is artificial. For example, B-cell chronic lymphocytic leukemia (CLL) and B-cell small lymphocytic lymphoma are simply different manifestations of the same neoplasm, as are lymphoblastic lymphomas and acute lymphocytic leukemias. Within the B-cell and T-cell categories, two subdivisions are recognized: precursor neoplasms, which correspond to the earliest stages of differentiation, and more mature differentiated neoplasms.[9,10]
Zeppa P, Marino G, Troncone G, et al.: Fine-needle cytology and flow cytometry immunophenotyping and subclassification of non-Hodgkin lymphoma: a critical review of 307 cases with technical suggestions. Cancer 102 (1): 55-65, 2004. [PUBMED Abstract]
Young NA, Al-Saleem T: Diagnosis of lymphoma by fine-needle aspiration cytology using the revised European-American classification of lymphoid neoplasms. Cancer 87 (6): 325-45, 1999. [PUBMED Abstract]
National Cancer Institute sponsored study of classifications of non-Hodgkin’s lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin’s Lymphoma Pathologic Classification Project. Cancer 49 (10): 2112-35, 1982. [PUBMED Abstract]
Pugh WC: Is the working formulation adequate for the classification of the low grade lymphomas? Leuk Lymphoma 10 (Suppl 1): 1-8, 1993.
Harris NL, Jaffe ES, Stein H, et al.: A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 84 (5): 1361-92, 1994. [PUBMED Abstract]
Pittaluga S, Bijnens L, Teodorovic I, et al.: Clinical analysis of 670 cases in two trials of the European Organization for the Research and Treatment of Cancer Lymphoma Cooperative Group subtyped according to the Revised European-American Classification of Lymphoid Neoplasms: a comparison with the Working Formulation. Blood 87 (10): 4358-67, 1996. [PUBMED Abstract]
Armitage JO, Weisenburger DD: New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 16 (8): 2780-95, 1998. [PUBMED Abstract]
A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 89 (11): 3909-18, 1997. [PUBMED Abstract]
Pileri SA, Milani M, Fraternali-Orcioni G, et al.: From the R.E.A.L. Classification to the upcoming WHO scheme: a step toward universal categorization of lymphoma entities? Ann Oncol 9 (6): 607-12, 1998. [PUBMED Abstract]
Society for Hematopathology Program: Society for Hematopathology Program. Am J Surg Pathol 21 (1): 114-121, 1997.
Stage Information for Indolent B-Cell Non-Hodgkin Lymphoma
Stage is important in selecting a treatment for patients with non-Hodgkin lymphoma (NHL). Chest and abdominal computed tomography (CT) scans are usually part of the staging evaluation for all patients with lymphoma. The staging system for NHL is similar to the staging system used for Hodgkin lymphoma (HL).
It is common for patients with NHL to have involvement of the following sites:
Noncontiguous lymph nodes.
Waldeyer ring.
Epitrochlear nodes.
Gastrointestinal tract.
Extranodal presentations. (A single extranodal site is occasionally the only site of involvement in patients with diffuse lymphoma.)
Bone marrow.
Liver (especially common in patients with low-grade lymphomas).
Cytological examination of cerebrospinal fluid may be positive in patients with aggressive NHL. Involvement of hilar and mediastinal lymph nodes is less common than in HL. Mediastinal adenopathy, however, is a prominent feature of lymphoblastic lymphoma and primary mediastinal B-cell lymphoma, entities primarily found in young adults.
Most patients with NHL present with advanced (stage III or stage IV) disease often identified by CT scans or biopsies of the bone marrow and other accessible sites of involvement. In a retrospective review of over 32,000 cases of lymphoma in France, up to 40% of diagnoses were made by core needle biopsy, and 60% were made by excisional biopsy.[1] After expert review, core needle biopsy provided a definite diagnosis in 92.3% of cases; excisional biopsy provided a definite diagnosis in 98.1% of cases (P < .0001). Laparoscopic biopsy or laparotomy is not required for staging but rarely may be necessary to establish a diagnosis or histological type.[2]
Positron emission tomography (PET) with fluorine F 18-fludeoxyglucose can be used for initial staging. It can also be used for follow-up after therapy as a supplement to CT scanning.[3] Multiple studies have demonstrated that interim PET scans after two to four cycles of therapy do not provide reliable prognostic information. A large cooperative group trial (ECOG-E344 [NCT00274924]) reported problems with interobserver reproducibility. Two prospective trials and one meta-analysis showed no differences in outcomes between PET-negative and PET-positive/biopsy-negative patients.[4–7]
In a retrospective study of 130 patients with diffuse large B-cell lymphoma, PET scanning identified all clinically important marrow involvement from lymphoma, and bone marrow biopsy did not upstage any patient’s lymphoma.[8] A retrospective study of 580 patients with follicular lymphoma from seven National Cancer Institute–sponsored trials showed no improvement in assessing response to therapy when bone marrow biopsy was added to radiological imaging.[9] The workup of NHL should include bone marrow biopsy when management would change (e.g., determining limited stage vs. advanced stage) or when evaluating cytopenias.
For patients with follicular lymphoma, a positive PET result after therapy has a worse prognosis; however, it is unclear whether a positive PET result is predictive when further or different therapy is implemented.[10]
Staging Subclassification System
Lugano classification
The American Joint Committee on Cancer (AJCC) has adopted the Lugano classification to evaluate and stage lymphoma.[11] The Lugano classification system replaces the Ann Arbor classification system, which was adopted in 1971 at the Ann Arbor Conference,[12] with some modifications 18 years later from the Cotswolds meeting.[13,14]
Table 2. Lugano Classification for Hodgkin and Non-Hodgkin Lymphomaa
Stage
Stage Description
Illustration
CSF = cerebrospinal fluid; CT = computed tomography; DLBCL = diffuse large B-cell lymphoma; NHL = non-Hodgkin lymphoma.
aHodgkin and Non-Hodgkin Lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 937–58.
bStage II bulky may be considered either early or advanced stage based on lymphoma histology and prognostic factors.
cThe definition of disease bulk varies according to lymphoma histology. In the Lugano classification, bulk ln Hodgkin lymphoma is defined as a mass greater than one-third of the thoracic diameter on CT of the chest or a mass >10 cm. For NHL, the recommended definitions of bulk vary by lymphoma histology. In follicular lymphoma, 6 cm has been suggested based on the Follicular Lymphoma International Prognostic Index-2 and its validation. In DLBCL, cutoffs ranging from 5 cm to 10 cm have been used, although 10 cm is recommended.
Limited stage
I
Involvement of a single lymphatic site (i.e., nodal region, Waldeyer’s ring, thymus, or spleen).
Diffuse or disseminated involvement of one or more extralymphatic organs, with or without associated lymph node involvement; or noncontiguous extralymphatic organ involvement in conjunction with nodal stage II disease; or any extralymphatic organ involvement in nodal stage III disease. Stage IV includes any involvement of the CSF, bone marrow, liver, or multiple lung lesions (other than by direct extension in stage IIE disease).
Note: Hodgkin lymphoma uses A or B designation with stage group. A/B is no longer used in NHL.
Occasionally, specialized staging systems are used. The physician should be aware of the system used in a specific report.
The E designation is used when extranodal lymphoid malignancies arise in tissues separate from, but near, the major lymphatic aggregates. Stage IV refers to disease that is diffusely spread throughout an extranodal site, such as the liver. If pathological proof of involvement of one or more extralymphatic sites has been documented, the symbol for the site of involvement, followed by a plus sign (+), is listed.
Table 3. Notation to Identify Specific Sites
N = nodes
H = liver
L = lung
M = bone marrow
S = spleen
P = pleura
O = bone
D = skin
Current practice assigns a clinical stage based on the findings of the clinical evaluation and a pathological stage based on the findings from invasive procedures beyond the initial biopsy.
For example, on percutaneous biopsy, a patient with inguinal adenopathy and a positive lymphangiogram without systemic symptoms might have involvement of the liver and bone marrow. The precise stage of such a patient would be clinical stage IIA, pathological stage IVA(H+)(M+).
Several other factors that are not included in the above staging system are important for the staging and prognosis of patients with NHL. These factors include the following:
Age.
Performance status (PS).
Tumor size.
Lactate dehydrogenase (LDH) values.
The number of extranodal sites.
References
Syrykh C, Chaouat C, Poullot E, et al.: Lymph node excisions provide more precise lymphoma diagnoses than core biopsies: a French Lymphopath network survey. Blood 140 (24): 2573-2583, 2022. [PUBMED Abstract]
Mann GB, Conlon KC, LaQuaglia M, et al.: Emerging role of laparoscopy in the diagnosis of lymphoma. J Clin Oncol 16 (5): 1909-15, 1998. [PUBMED Abstract]
Barrington SF, Mikhaeel NG, Kostakoglu L, et al.: Role of imaging in the staging and response assessment of lymphoma: consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 32 (27): 3048-58, 2014. [PUBMED Abstract]
Horning SJ, Juweid ME, Schöder H, et al.: Interim positron emission tomography scans in diffuse large B-cell lymphoma: an independent expert nuclear medicine evaluation of the Eastern Cooperative Oncology Group E3404 study. Blood 115 (4): 775-7; quiz 918, 2010. [PUBMED Abstract]
Moskowitz CH, Schöder H, Teruya-Feldstein J, et al.: Risk-adapted dose-dense immunochemotherapy determined by interim FDG-PET in Advanced-stage diffuse large B-Cell lymphoma. J Clin Oncol 28 (11): 1896-903, 2010. [PUBMED Abstract]
Pregno P, Chiappella A, Bellò M, et al.: Interim 18-FDG-PET/CT failed to predict the outcome in diffuse large B-cell lymphoma patients treated at the diagnosis with rituximab-CHOP. Blood 119 (9): 2066-73, 2012. [PUBMED Abstract]
Sun N, Zhao J, Qiao W, et al.: Predictive value of interim PET/CT in DLBCL treated with R-CHOP: meta-analysis. Biomed Res Int 2015: 648572, 2015. [PUBMED Abstract]
Khan AB, Barrington SF, Mikhaeel NG, et al.: PET-CT staging of DLBCL accurately identifies and provides new insight into the clinical significance of bone marrow involvement. Blood 122 (1): 61-7, 2013. [PUBMED Abstract]
Rutherford SC, Yin J, Pederson L, et al.: Relevance of Bone Marrow Biopsies for Response Assessment in US National Cancer Institute National Clinical Trials Network Follicular Lymphoma Clinical Trials. J Clin Oncol 41 (2): 336-342, 2023. [PUBMED Abstract]
Pyo J, Won Kim K, Jacene HA, et al.: End-therapy positron emission tomography for treatment response assessment in follicular lymphoma: a systematic review and meta-analysis. Clin Cancer Res 19 (23): 6566-77, 2013. [PUBMED Abstract]
Hodgkin and non-Hodgkin lymphoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 937–58.
Carbone PP, Kaplan HS, Musshoff K, et al.: Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res 31 (11): 1860-1, 1971. [PUBMED Abstract]
Lister TA, Crowther D, Sutcliffe SB, et al.: Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol 7 (11): 1630-6, 1989. [PUBMED Abstract]
National Cancer Institute sponsored study of classifications of non-Hodgkin’s lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin’s Lymphoma Pathologic Classification Project. Cancer 49 (10): 2112-35, 1982. [PUBMED Abstract]
Indolent B-Cell Non-Hodgkin Lymphoma
Indolent B-cell non-Hodgkin lymphoma (NHL) includes the following subtypes:
Follicular lymphoma makes up 20% of all NHL and as many as 70% of the indolent lymphomas reported in American and European clinical trials.[1–3] Most patients with follicular lymphoma are aged 50 years and older and present with widespread disease at diagnosis. Nodal involvement is most common and is often accompanied by splenic and bone marrow disease. Rearrangement of the BCL2 gene is present in more than 90% of patients with follicular lymphoma. Overexpression of the BCL2 protein is associated with the inability to eradicate the lymphoma by inhibiting apoptosis.[4]
Prognosis
Follicular lymphoma is designated as indolent because median survival ranges from 8 to 15 years, even in advanced stages.[5–7] Patients with advanced-stage follicular lymphoma are not cured with current therapeutic options. The rate of relapse is fairly consistent over time, even in patients who have achieved complete responses to treatment.[8] Watchful waiting, the deferring of treatment until the patient becomes symptomatic, is an option for patients with advanced-stage follicular lymphoma.[9,10] An international index for follicular lymphoma (the Follicular Lymphoma International Prognostic Index [FLIPI]) [11–13] identified five significant risk factors prognostic of overall survival (OS):
Age (≤60 years vs. >60 years).
Serum lactate dehydrogenase (LDH) (normal vs. elevated).
Stage (stage I or stage II vs. stage III or stage IV).
Hemoglobin level (≥120 g/L vs. <120 g/L).
Number of nodal areas (≤4 vs. >4).
Patients with zero or one risk factor have a 10-year survival rate of 67%, and four or five risk factors confer a 10-year survival rate of 36%.[11] In a revised FLIPI-2, an elevated beta-2-microglobulin and lymph node size of more than 6 cm are proposed prognostic factors instead of serum LDH and the number of nodal areas.[14] Although the FLIPI and FLIPI-2 indices can predict progression-free survival (PFS) and OS, the scores cannot be used to establish the need for therapy or to predict response to therapy.[11,14] The primary use of FLIPI or FLIPI-2 is to assure a balance of prognostic factors or to define entry requirements in randomized clinical trials. Individuals with an adverse FLIPI score may well benefit from watchful waiting or may respond well to initial therapy. An alternative prognostic index using only beta-2-microglobulin and initial bone marrow involvement (PRIMA-PI) has the disadvantage of requiring an invasive test not usually required outside the context of a clinical trial.[15] The Follicular Lymphoma Evaluation Index (FLEX), an alternative prognostic index using only noninvasive clinical variables, outperformed FLIPI, FLIPI-2, and PRIMA-PI, using data from immunochemotherapy trials.[16] The nine clinical variables of the FLEX model include:
Male sex.
Histological grade 3a disease.
More than two extranodal sites.
Eastern Cooperative Oncology Group performance status score of 2 or higher.
Hemoglobin level less than 120 g/L.
Beta-2 microglobulin level above the upper limit of normal.
LDH level above the upper limit of normal.
Absolute blood natural killer cell count less than 100/μL.
Bulky disease.
Three retrospective analyses, including one pooled analysis of 5,225 patients from 13 randomized clinical trials, identified a high-risk group that had a 5-year OS rate of 50% when relapses occurred within 24 months of induction chemoimmunotherapy.[17–19] A fourth retrospective analysis of 296 patients who received bendamustine and rituximab found a 2-year OS rate of 38% (95% confidence interval [CI], 20%−55%) among those with progression of disease before 24 months (POD24). Most of these patients (76%) had transformed disease (histological progression to diffuse large B-cell lymphoma [DLBCL]).[20] These patients with higher-risk POD24 disease represent a target population for clinical trials.
Therapeutic approaches
Because of the often-indolent clinical course and the lack of symptoms in some patients with follicular lymphoma, watchful waiting remains a standard of care during the initial encounter and for patients with slow asymptomatic relapsing disease. When therapy is required, numerous options may be used in varying sequences with an OS equivalence at 5 to 10 years.[9,21–23] Rituximab can be given alone or in combination with various chemotherapy options.[23–25] Rituximab can also be combined with the immunomodulating-agent lenalidomide to avoid the short- and long-term toxicities of cytotoxic agents.[26–28] Obinutuzumab is a completely humanized anti–CD20 monoclonal antibody that can be given alone or with combination chemotherapy. It may be especially useful for patients who develop severe allergic reactions to rituximab due to human antimouse antibodies.[29] CD19-directed chimeric antigen receptor T cells may be used in patients who have disease progression after two or more prior lines of therapy.[30] Mosunetuzumab, a bispecific CD20-directed CD3 T-cell engager, may also be used in this setting.[31] Consolidation therapy for relapsed disease after reinduction therapy using autologous stem cell transplant (SCT) or allogeneic SCT can be considered.[32]
Outside the context of clinical trials, the use of measurable residual disease (MRD) testing has not been shown to be predictive in directing therapy for patients with follicular lymphoma. In retrospective analyses of two randomized prospective trials, while MRD negativity was prognostic of outcome, maintenance rituximab or obinutuzumab prolonged PFS the most among patients with MRD-negative disease.[33,34][Level of evidence C2] Stopping maintenance rituximab or obinutuzumab was not indicated in patients with MRD-negative disease, negating any possible change in therapy based on that status.
Follicular lymphoma in situ and primary follicular lymphoma of the duodenum are particularly indolent variants that rarely progress or require therapy.[35,36] A so-called pediatric-type nodal follicular lymphoma has indolent behavior and rarely recurs; adult patients with this histological variant have disease characterized by a lack of BCL2 rearrangement in conjunction with a Ki-67 proliferation index greater than 30% and a localized stage I presentation.[37]
Patients with indolent lymphoma may experience a relapse with a more aggressive histology. If the clinical pattern of relapse suggests that the disease is behaving in a more aggressive manner, a biopsy can be performed, if feasible.[38] If disease conversion to a more aggressive histology is confirmed, therapy must change to a regimen applicable to that histological type.[39] Rapid growth or discordant growth between various disease sites may indicate a histological conversion.[38]
In a prospective nonrandomized study, at a median follow-up of 6.8 years, 379 of 2,652 patients (14%) subsequently transformed to a more aggressive histology after an initial diagnosis of follicular lymphoma.[40][Level of evidence C3] The median OS after subsequent transformation was 5 years. However, among 47 patients with evidence of transformation in conjunction with follicular lymphoma at the time of initial diagnosis, the OS was no worse than that of the nontransformed patients (5-year OS rate, 88%; 95% CI, 74%–95%).
Lymphoplasmacytic lymphoma is usually associated with a monoclonal serum paraprotein of immunoglobulin M (IgM) type (Waldenström macroglobulinemia).[41] Most patients have bone marrow, lymph node, and splenic involvement, and some patients may develop hyperviscosity syndrome. Most patients with Waldenström macroglobulinemia carry the MYD88 variant, which some pathologists consider indicative for the disease.[42] Other lymphomas may also be associated with serum paraproteins. Patients with lymphoplasmacytic lymphoma should be checked for associated hepatitis C virus (HCV) infection.
Asymptomatic patients can be monitored for evidence of disease progression without immediate need for chemotherapy.[9,43,44]
Prognostic factors associated with symptoms requiring therapy include:
An externally validated prognostic model uses age, albumin, and LDH levels.[45]
Therapeutic approaches
The management of lymphoplasmacytic lymphoma is similar to that of other low-grade lymphomas, especially diffuse small lymphocytic lymphoma/chronic lymphocytic leukemia.[46–48] If the serum viscosity relative to water is greater than four, the patient may have symptoms of hyperviscosity. Plasmapheresis is useful for temporary, acute symptoms such as retinopathy, congestive heart failure, and central nervous system (CNS) dysfunction. It can also be combined with chemotherapy for prolonged disease control. Symptomatic patients with a serum viscosity of four or lower are usually treated with chemoimmunotherapy or biologically directed therapies. Therapy may be required to correct hemolytic anemia in patients with chronic cold agglutinin disease; rituximab, bendamustine, and steroids are often used.[44] Occasionally, a heated room is required for patients whose cold agglutinins become activated by even minor chilling. Sutimlimab, an immunoglobulin G4 monoclonal antibody that selectively inhibits the complement pathway at C15, can reduce hemolysis when therapies directed at the lymphoplasmacytic lymphoma are ineffective.[49]
First-line regimens include zanubrutinib (a Bruton tyrosine kinase [BTK] inhibitor), rituximab, and ibrutinib (another BTK inhibitor), rituximab alone, the nucleoside analogues, and alkylating agents, either as single agents or as part of combination chemotherapy.[50–53] In a randomized prospective trial, 150 symptomatic patients (including patients with previously untreated and relapsing disease) received either ibrutinib and rituximab or rituximab and a placebo. With a median follow-up of 50 months, the 4.5-year PFS rate was 68% in the ibrutinib-and-rituximab arm (95% CI, 55%–78%) and 25% in the rituximab-and-placebo arm (95% CI, 15%–37%) (hazard ratio [HR], 0.25; 95% CI, 0.15–0.42; P < .0001). The OS rate at 30 months was no different in the two arms (92%–94%).[52][Level of evidence B1] Zanubrutinib was compared with ibrutinib in a randomized prospective clinical trial of 164 patients with relapsed disease and 38 with previously untreated disease.[53] With a median follow-up of 44.4 months, the PFS rate was similar in both groups at 70% to 78% (HR, 0.63; 95% CI, 0.36–1.12), and the OS rate was similar in both groups at 85% to 87% (HR, 0.75; 95% CI, 0.36–1.59).[54] The zanubrutinib group had fewer cases of atrial fibrillation (11 vs. 1) and 50% fewer cases of hypertension (statistics not provided).[53][Level of evidence C3] BTK inhibition with ibrutinib allowed all 13 patients with cold-antibody–mediated autoimmune hemolytic anemia and acrocyanosis to attain clinical remission regardless of underlying pathology or MYD88 variant status.[55][Level of evidence C3]
Previously untreated patients who received rituximab had response rates of 60% to 80%, but close monitoring of the serum IgM is required because of a sudden rise in this paraprotein at the start of therapy.[56–58][Level of evidence C3] The rise of IgM after rituximab can be avoided with the concomitant use of an alkylating agent, such as cyclophosphamide, or the proteosome inhibitors bortezomib or ixazomib.[44,59–61] A combination of bortezomib, dexamethasone, and rituximab has been used without causing IgM rebound.[62–64] Previously untreated patients with lymphoplasmacytic lymphoma who received the nucleoside analogues cladribine and fludarabine showed similar response rates.[51,65,66][Level of evidence C3] Patients who received single-agent alkylators, bendamustine, bortezomib, venetoclax, and combination chemotherapy with or without rituximab also showed similar response rates.[51,59,61,67–71][Level of evidence C3] In the rare case of lymphoplasmacytic lymphoma involving the CNS (Bing-Neel syndrome), ibrutinib resulted in an 85% response rate in an anecdotal series of 28 patients.[72][Level of evidence C3]
Myeloablative therapy with autologous or allogeneic hematopoietic stem cell support is under clinical evaluation.[73–76] Candidates for this approach should avoid long-term use of alkylating agents or purine nucleoside analogues, which can deplete hematopoietic stem cells or predispose patients to myelodysplasia or acute leukemia.[56,77] After relapse from alkylating-agent therapy, 92 patients with lymphoplasmacytic lymphoma were randomly assigned to receive either fludarabine or cyclophosphamide, doxorubicin, and prednisone. Although relapse-free survival favored fludarabine (median duration of 19 months vs. 3 months; P < .01), no difference was observed in OS.[78][Level of evidence B1]
Marginal Zone Lymphoma
When marginal zone lymphomas involve the nodes, they are called monocytoid B-cell lymphomas or nodal marginal zone B-cell lymphomas. When they involve extranodal sites (e.g., gastrointestinal tract, thyroid, lung, breast, orbit, and skin), they are called mucosa-associated lymphatic tissue (MALT) lymphomas.[79,80] Splenic marginal zone lymphoma is a distinct clinical entity, which usually presents with massive splenomegaly. A variant form of MALT lymphoma is known as immunoproliferative small intestinal disease (IPSID).[80] A prognostic index for all of the marginal zone lymphomas has three adverse prognostic factors: aged 70 years or older, stage III or stage IV disease, and high LDH level.[81] Fewer than 10% of patients transform to a higher-grade lymphoma. In one retrospective review, risk factors for transformation included elevated LDH, more than four nodal sites at the time of initial diagnosis of marginal zone lymphoma, and failure to achieve complete response after initial treatment.[82]
Gastric MALT
Many patients have a history of autoimmune disease, such as Hashimoto thyroiditis or Sjögren syndrome, or of Helicobacter gastritis. Most patients present with stage I or stage II extranodal disease, which is most often in the stomach. Treatment of H. pylori infection may resolve most cases of localized gastric involvement.[83,84] After standard antibiotic regimens, 50% of patients show resolution of gastric MALT by endoscopy after 3 months. Other patients may show resolution after 12 to 18 months of observation. Of the patients who attain complete remission, 30% demonstrate monoclonality by immunoglobulin heavy chain rearrangement on stomach biopsies with a 5-year median follow-up.[85] The clinical implication of this finding is unknown. Translocation t(11;18) in patients with gastric MALT predicts poor response to both antibiotic therapy and oral alkylator therapy, and predicts negative H. pylori testing results.[86–88] Patients with stable asymptomatic disease and persistently positive biopsies have been successfully followed with a watchful waiting approach until disease progression.[84] Patients with disease progression are treated with radiation therapy,[89–94] rituximab,[95] surgery (total gastrectomy or partial gastrectomy plus radiation therapy),[96] chemotherapy,[97] or combined-modality therapy.[98] A single-arm prospective trial enrolled 24 patients with newly diagnosed or relapsed H. pylori–negative gastric MALT.[94] Most patients had a complete response to radiation therapy at 4 Gy, and 20 Gy was applied as salvage therapy. The 3-year local control rate was 96% (95% CI, 88%–100%).[94][Level of evidence C3] Endoscopic ultrasonography may help clinicians monitor responses in these patients.[99] Four case series encompassing more than 100 patients with stage IE or IIE DLBCL with or without associated MALT (but H. pylori-positive) reported durable complete remissions in more than 50% of the patients after treatment of H. pylori.[100–103]
Extragastric MALT
Localized involvement of other sites can be treated with radiation therapy or surgery.[90–92,104–107] Patients with extragastric MALT lymphoma have a higher relapse rate than patients with gastric MALT lymphoma in some series, and relapses can happen many years and even decades later.[108] Many of these recurrences involve different MALT sites than the original location.[109] When disseminated to lymph nodes, bone marrow, or blood, this entity behaves like other low-grade lymphomas.[110,111] A prospective randomized trial of 401 patients with nongastric extranodal MALT compared chlorambucil alone versus rituximab plus chlorambucil versus rituximab alone.[112] With a median follow-up of 7.4 years, the event-free survival was 68% in the rituximab-plus-chlorambucil arm, 51% in the rituximab-alone arm, and 50% in the chlorambucil-alone arm (P = .0009). However, the 5-year OS rate was 90% in all arms.[112] For patients with ocular adnexal MALT, antibiotic therapy using doxycycline that targeted Chlamydia psittaci resulted in durable remissions for almost one-half of the patients in a review of the literature that included 131 patients.[113][Level of evidence C3] These responses to doxycycline are mainly seen in Italian trials and less often in trials conducted in other geographic sites.[114] Large B-cell lymphomas of MALT sites are classified and treated as diffuse large cell lymphomas.[115] A large, retrospective review of primary ocular adnexal MALT found that after 10 years of follow-up, 4% of stage I patients treated with radiation therapy had disease transformation to DLBCL, and 3% developed CNS involvement.[116]
Nodal marginal zone lymphoma
Patients with nodal marginal zone lymphoma (monocytoid B-cell lymphoma) are treated with watchful waiting or therapies as described for lymphoplasmacytic lymphoma. Rituximab alone, obinutuzumab alone, or combinations with cytotoxic agents (such as bendamustine, CVP [cyclophosphamide, vincristine, and prednisone] or CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone]) can be used.[117–119][Level of evidence C3] Zanubrutinib is approved for patients with disease relapse after a rituximab-containing regimen. This approval was based on a single-arm phase II study.[120] With a median follow-up of 15.7 months, the overall response rate was 68.2%, and the complete response rate was 25.8%. The median duration of response was 93% at 12 months.[120][Level of evidence C3] Ibrutinib also showed similar efficacy in patients with relapsed marginal zone lymphoma.[121][Level of evidence C3] Patients with marginal zone lymphoma comprised a small percentage (about 15%) of the population for a trial that established the benefit of rituximab with lenalidomide in patients with relapsed or refractory indolent lymphoma (AUGMENT trial).[122][Level of evidence C3] Similar to follicular lymphoma, patients with POD24 who required initiation of therapy had a worse prognosis than did the patients without POD24 (3-year OS rate, 53% vs. 95%).[123] Among patients with concomitant HCV infection, 40% to 60% attained a complete or partial remission after loss of detectable HCV RNA with antiviral treatment.[124,125][Level of evidence C3]
Mediterranean abdominal lymphoma
The disease variously known as Mediterranean abdominal lymphoma, heavy-chain disease, or IPSID, which occurs in young adults in eastern Mediterranean countries, is another version of MALT lymphoma. This disease responds to antibiotics in its early stages.[126] Campylobacter jejuni has been identified as one of the bacterial species associated with IPSID, and antibiotic therapy may result in disease remission.[127]
Splenic marginal zone lymphoma
Splenic marginal zone lymphoma is an indolent lymphoma that is marked by massive splenomegaly and peripheral blood and bone marrow involvement, usually without adenopathy.[128,129] This type of lymphoma is also known as splenic lymphoma with villous lymphocytes. Splenectomy may result in prolonged remission.[130,131]
Management is similar to that of other low-grade lymphomas and usually involves rituximab alone or rituximab in combination with purine analogues or alkylating agent chemotherapy.[132] Splenic marginal zone lymphoma responds less well to chemotherapy, which would ordinarily be effective for chronic lymphocytic leukemia.[128,132,133] Among small numbers of patients with splenic marginal zone lymphoma (splenic lymphoma with villous lymphocytes) and HCV infection, most attained a complete or partial remission after loss of detectable HCV RNA with treatment using interferon alfa with or without ribavirin.[124,134]; [135][Level of evidence C3] In contrast, no responses to interferon were seen in six HCV-negative patients.
References
Armitage JO, Weisenburger DD: New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 16 (8): 2780-95, 1998. [PUBMED Abstract]
A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 89 (11): 3909-18, 1997. [PUBMED Abstract]
Society for Hematopathology Program: Society for Hematopathology Program. Am J Surg Pathol 21 (1): 114-121, 1997.
López-Guillermo A, Cabanillas F, McDonnell TI, et al.: Correlation of bcl-2 rearrangement with clinical characteristics and outcome in indolent follicular lymphoma. Blood 93 (9): 3081-7, 1999. [PUBMED Abstract]
Peterson BA, Petroni GR, Frizzera G, et al.: Prolonged single-agent versus combination chemotherapy in indolent follicular lymphomas: a study of the cancer and leukemia group B. J Clin Oncol 21 (1): 5-15, 2003. [PUBMED Abstract]
Swenson WT, Wooldridge JE, Lynch CF, et al.: Improved survival of follicular lymphoma patients in the United States. J Clin Oncol 23 (22): 5019-26, 2005. [PUBMED Abstract]
Liu Q, Fayad L, Cabanillas F, et al.: Improvement of overall and failure-free survival in stage IV follicular lymphoma: 25 years of treatment experience at The University of Texas M.D. Anderson Cancer Center. J Clin Oncol 24 (10): 1582-9, 2006. [PUBMED Abstract]
Ardeshna KM, Smith P, Norton A, et al.: Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a randomised controlled trial. Lancet 362 (9383): 516-22, 2003. [PUBMED Abstract]
Armitage JO, Longo DL: Is watch and wait still acceptable for patients with low-grade follicular lymphoma? Blood 127 (23): 2804-8, 2016. [PUBMED Abstract]
Solal-Céligny P, Roy P, Colombat P, et al.: Follicular lymphoma international prognostic index. Blood 104 (5): 1258-65, 2004. [PUBMED Abstract]
Perea G, Altés A, Montoto S, et al.: Prognostic indexes in follicular lymphoma: a comparison of different prognostic systems. Ann Oncol 16 (9): 1508-13, 2005. [PUBMED Abstract]
Buske C, Hoster E, Dreyling M, et al.: The Follicular Lymphoma International Prognostic Index (FLIPI) separates high-risk from intermediate- or low-risk patients with advanced-stage follicular lymphoma treated front-line with rituximab and the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) with respect to treatment outcome. Blood 108 (5): 1504-8, 2006. [PUBMED Abstract]
Federico M, Bellei M, Marcheselli L, et al.: Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol 27 (27): 4555-62, 2009. [PUBMED Abstract]
Bachy E, Maurer MJ, Habermann TM, et al.: A simplified scoring system in de novo follicular lymphoma treated initially with immunochemotherapy. Blood 132 (1): 49-58, 2018. [PUBMED Abstract]
Mir F, Mattiello F, Grigg A, et al.: Follicular Lymphoma Evaluation Index (FLEX): A new clinical prognostic model that is superior to existing risk scores for predicting progression-free survival and early treatment failure after frontline immunochemotherapy. Am J Hematol 95 (12): 1503-1510, 2020. [PUBMED Abstract]
Casulo C, Byrtek M, Dawson KL, et al.: Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J Clin Oncol 33 (23): 2516-22, 2015. [PUBMED Abstract]
Shi Q, Flowers CR, Hiddemann W, et al.: Thirty-Month Complete Response as a Surrogate End Point in First-Line Follicular Lymphoma Therapy: An Individual Patient-Level Analysis of Multiple Randomized Trials. J Clin Oncol 35 (5): 552-560, 2017. [PUBMED Abstract]
Casulo C, Dixon JG, Le-Rademacher J, et al.: Validation of POD24 as a robust early clinical end point of poor survival in FL from 5225 patients on 13 clinical trials. Blood 139 (11): 1684-1693, 2022. [PUBMED Abstract]
Freeman CL, Kridel R, Moccia AA, et al.: Early progression after bendamustine-rituximab is associated with high risk of transformation in advanced stage follicular lymphoma. Blood 134 (9): 761-764, 2019. [PUBMED Abstract]
Brice P, Bastion Y, Lepage E, et al.: Comparison in low-tumor-burden follicular lymphomas between an initial no-treatment policy, prednimustine, or interferon alfa: a randomized study from the Groupe d’Etude des Lymphomes Folliculaires. Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol 15 (3): 1110-7, 1997. [PUBMED Abstract]
Young RC, Longo DL, Glatstein E, et al.: The treatment of indolent lymphomas: watchful waiting v aggressive combined modality treatment. Semin Hematol 25 (2 Suppl 2): 11-6, 1988. [PUBMED Abstract]
Luminari S, Ferrari A, Manni M, et al.: Long-Term Results of the FOLL05 Trial Comparing R-CVP Versus R-CHOP Versus R-FM for the Initial Treatment of Patients With Advanced-Stage Symptomatic Follicular Lymphoma. J Clin Oncol 36 (7): 689-696, 2018. [PUBMED Abstract]
Lockmer S, Østenstad B, Hagberg H, et al.: Chemotherapy-Free Initial Treatment of Advanced Indolent Lymphoma Has Durable Effect With Low Toxicity: Results From Two Nordic Lymphoma Group Trials With More Than 10 Years of Follow-Up. J Clin Oncol : JCO1800262, 2018. [PUBMED Abstract]
Cartron G, Bachy E, Tilly H, et al.: Randomized Phase III Trial Evaluating Subcutaneous Rituximab for the First-Line Treatment of Low-Tumor Burden Follicular Lymphoma: Results of a LYSA Study. J Clin Oncol 41 (19): 3523-3533, 2023. [PUBMED Abstract]
Morschhauser F, Fowler NH, Feugier P, et al.: Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N Engl J Med 379 (10): 934-947, 2018. [PUBMED Abstract]
Leonard JP, Trnený M, Izutsu K, et al.: Augment: a phase III randomized study of lenalidomide Plus rituximab (R2) vs rituximab/placebo in patients with relapsed/refractory indolent non-Hodgkin lymphoma. [Abstract] Blood 132 (Suppl 1): A-445, 2018.
Zucca E, Rondeau S, Vanazzi A, et al.: Short regimen of rituximab plus lenalidomide in follicular lymphoma patients in need of first-line therapy. Blood 134 (4): 353-362, 2019. [PUBMED Abstract]
Marcus R, Davies A, Ando K, et al.: Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N Engl J Med 377 (14): 1331-1344, 2017. [PUBMED Abstract]
Jacobson CA, Chavez JC, Sehgal AR, et al.: Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 23 (1): 91-103, 2022. [PUBMED Abstract]
Bartlett NL, Sehn LH, Matasar MJ, et al.: Mosunetuzumab monotherapy demonstrates durable efficacy with a manageable safety profile in patients with relapsed/refractory follicular lymphoma who received ≥2 prior therapies: updated results from a pivotal phase II study. [Abstract] Blood 140 (Suppl 1): A-610, 1467-70, 2022.
Schaaf M, Reiser M, Borchmann P, et al.: High-dose therapy with autologous stem cell transplantation versus chemotherapy or immuno-chemotherapy for follicular lymphoma in adults. Cochrane Database Syst Rev 1: CD007678, 2012. [PUBMED Abstract]
Luminari S, Manni M, Galimberti S, et al.: Response-Adapted Postinduction Strategy in Patients With Advanced-Stage Follicular Lymphoma: The FOLL12 Study. J Clin Oncol 40 (7): 729-739, 2022. [PUBMED Abstract]
Pott C, Jurinovic V, Trotman J, et al.: Minimal Residual Disease Status Predicts Outcome in Patients With Previously Untreated Follicular Lymphoma: A Prospective Analysis of the Phase III GALLIUM Study. J Clin Oncol 42 (5): 550-561, 2024. [PUBMED Abstract]
Schmatz AI, Streubel B, Kretschmer-Chott E, et al.: Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol 29 (11): 1445-51, 2011. [PUBMED Abstract]
Jegalian AG, Eberle FC, Pack SD, et al.: Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood 118 (11): 2976-84, 2011. [PUBMED Abstract]
Louissaint A, Ackerman AM, Dias-Santagata D, et al.: Pediatric-type nodal follicular lymphoma: an indolent clonal proliferation in children and adults with high proliferation index and no BCL2 rearrangement. Blood 120 (12): 2395-404, 2012. [PUBMED Abstract]
Sarkozy C, Trneny M, Xerri L, et al.: Risk Factors and Outcomes for Patients With Follicular Lymphoma Who Had Histologic Transformation After Response to First-Line Immunochemotherapy in the PRIMA Trial. J Clin Oncol 34 (22): 2575-82, 2016. [PUBMED Abstract]
Tsimberidou AM, O’Brien S, Khouri I, et al.: Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol 24 (15): 2343-51, 2006. [PUBMED Abstract]
Wagner-Johnston ND, Link BK, Byrtek M, et al.: Outcomes of transformed follicular lymphoma in the modern era: a report from the National LymphoCare Study (NLCS). Blood 126 (7): 851-7, 2015. [PUBMED Abstract]
Leblond V, Kastritis E, Advani R, et al.: Treatment recommendations from the Eighth International Workshop on Waldenström’s Macroglobulinemia. Blood 128 (10): 1321-8, 2016. [PUBMED Abstract]
Treon SP, Xu L, Yang G, et al.: MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 367 (9): 826-33, 2012. [PUBMED Abstract]
Dhodapkar MV, Hoering A, Gertz MA, et al.: Long-term survival in Waldenstrom macroglobulinemia: 10-year follow-up of Southwest Oncology Group-directed intergroup trial S9003. Blood 113 (4): 793-6, 2009. [PUBMED Abstract]
Ansell SM, Kyle RA, Reeder CB, et al.: Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin Proc 85 (9): 824-33, 2010. [PUBMED Abstract]
Zanwar S, Le-Rademacher J, Durot E, et al.: Simplified Risk Stratification Model for Patients With Waldenström Macroglobulinemia. J Clin Oncol 42 (21): 2527-2536, 2024. [PUBMED Abstract]
Kapoor P, Ansell SM, Fonseca R, et al.: Diagnosis and Management of Waldenström Macroglobulinemia: Mayo Stratification of Macroglobulinemia and Risk-Adapted Therapy (mSMART) Guidelines 2016. JAMA Oncol 3 (9): 1257-1265, 2017. [PUBMED Abstract]
Dimopoulos MA, Kastritis E: How I treat Waldenström macroglobulinemia. Blood 134 (23): 2022-2035, 2019. [PUBMED Abstract]
Gertz MA: Waldenstrom Macroglobulinemia: Tailoring Therapy for the Individual. J Clin Oncol 40 (23): 2600-2608, 2022. [PUBMED Abstract]
Röth A, Berentsen S, Barcellini W, et al.: Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial. Blood 140 (9): 980-991, 2022. [PUBMED Abstract]
Buske C, Dimopoulos MA, Grunenberg A, et al.: Bortezomib-Dexamethasone, Rituximab, and Cyclophosphamide as First-Line Treatment for Waldenström’s Macroglobulinemia: A Prospectively Randomized Trial of the European Consortium for Waldenström’s Macroglobulinemia. J Clin Oncol 41 (14): 2607-2616, 2023. [PUBMED Abstract]
Leblond V, Johnson S, Chevret S, et al.: Results of a randomized trial of chlorambucil versus fludarabine for patients with untreated Waldenström macroglobulinemia, marginal zone lymphoma, or lymphoplasmacytic lymphoma. J Clin Oncol 31 (3): 301-7, 2013. [PUBMED Abstract]
Buske C, Tedeschi A, Trotman J, et al.: Ibrutinib Plus Rituximab Versus Placebo Plus Rituximab for Waldenström’s Macroglobulinemia: Final Analysis From the Randomized Phase III iNNOVATE Study. J Clin Oncol 40 (1): 52-62, 2022. [PUBMED Abstract]
Tam CS, Opat S, D’Sa S, et al.: A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood 136 (18): 2038-2050, 2020. [PUBMED Abstract]
Dimopoulos MA, Opat S, D’Sa S, et al.: Zanubrutinib Versus Ibrutinib in Symptomatic Waldenström Macroglobulinemia: Final Analysis From the Randomized Phase III ASPEN Study. J Clin Oncol 41 (33): 5099-5106, 2023. [PUBMED Abstract]
Jalink M, Berentsen S, Castillo JJ, et al.: Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood 138 (20): 2002-2005, 2021. [PUBMED Abstract]
Gertz MA, Anagnostopoulos A, Anderson K, et al.: Treatment recommendations in Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol 30 (2): 121-6, 2003. [PUBMED Abstract]
Dimopoulos MA, Zervas C, Zomas A, et al.: Treatment of Waldenström’s macroglobulinemia with rituximab. J Clin Oncol 20 (9): 2327-33, 2002. [PUBMED Abstract]
Treon SP, Branagan AR, Hunter Z, et al.: Paradoxical increases in serum IgM and viscosity levels following rituximab in Waldenstrom’s macroglobulinemia. Ann Oncol 15 (10): 1481-3, 2004. [PUBMED Abstract]
Dimopoulos MA, Chen C, Kastritis E, et al.: Bortezomib as a treatment option in patients with Waldenström macroglobulinemia. Clin Lymphoma Myeloma Leuk 10 (2): 110-7, 2010. [PUBMED Abstract]
Gavriatopoulou M, García-Sanz R, Kastritis E, et al.: BDR in newly diagnosed patients with WM: final analysis of a phase 2 study after a minimum follow-up of 6 years. Blood 129 (4): 456-459, 2017. [PUBMED Abstract]
Kersten MJ, Amaador K, Minnema MC, et al.: Combining Ixazomib With Subcutaneous Rituximab and Dexamethasone in Relapsed or Refractory Waldenström’s Macroglobulinemia: Final Analysis of the Phase I/II HOVON124/ECWM-R2 Study. J Clin Oncol 40 (1): 40-51, 2022. [PUBMED Abstract]
Treon SP, Ioakimidis L, Soumerai JD, et al.: Primary therapy of Waldenström macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. J Clin Oncol 27 (23): 3830-5, 2009. [PUBMED Abstract]
Dimopoulos MA, García-Sanz R, Gavriatopoulou M, et al.: Primary therapy of Waldenstrom macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): long-term results of a phase 2 study of the European Myeloma Network (EMN). Blood 122 (19): 3276-82, 2013. [PUBMED Abstract]
Treon SP, Tripsas CK, Meid K, et al.: Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenström’s macroglobulinemia. Blood 124 (4): 503-10, 2014. [PUBMED Abstract]
Laszlo D, Andreola G, Rigacci L, et al.: Rituximab and subcutaneous 2-chloro-2′-deoxyadenosine combination treatment for patients with Waldenstrom macroglobulinemia: clinical and biologic results of a phase II multicenter study. J Clin Oncol 28 (13): 2233-8, 2010. [PUBMED Abstract]
García-Sanz R, Montoto S, Torrequebrada A, et al.: Waldenström macroglobulinaemia: presenting features and outcome in a series with 217 cases. Br J Haematol 115 (3): 575-82, 2001. [PUBMED Abstract]
Buske C, Hoster E, Dreyling M, et al.: The addition of rituximab to front-line therapy with CHOP (R-CHOP) results in a higher response rate and longer time to treatment failure in patients with lymphoplasmacytic lymphoma: results of a randomized trial of the German Low-Grade Lymphoma Study Group (GLSG). Leukemia 23 (1): 153-61, 2009. [PUBMED Abstract]
Ghobrial IM, Hong F, Padmanabhan S, et al.: Phase II trial of weekly bortezomib in combination with rituximab in relapsed or relapsed and refractory Waldenstrom macroglobulinemia. J Clin Oncol 28 (8): 1422-8, 2010. [PUBMED Abstract]
Rummel MJ, Niederle N, Maschmeyer G, et al.: Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381 (9873): 1203-10, 2013. [PUBMED Abstract]
Castillo JJ, Allan JN, Siddiqi T, et al.: Venetoclax in Previously Treated Waldenström Macroglobulinemia. J Clin Oncol 40 (1): 63-71, 2022. [PUBMED Abstract]
Castillo JJ, Itchaki G, Paludo J, et al.: Ibrutinib for the treatment of Bing-Neel syndrome: a multicenter study. Blood 133 (4): 299-305, 2019. [PUBMED Abstract]
Dreger P, Glass B, Kuse R, et al.: Myeloablative radiochemotherapy followed by reinfusion of purged autologous stem cells for Waldenström’s macroglobulinaemia. Br J Haematol 106 (1): 115-8, 1999. [PUBMED Abstract]
Desikan R, Dhodapkar M, Siegel D, et al.: High-dose therapy with autologous haemopoietic stem cell support for Waldenström’s macroglobulinaemia. Br J Haematol 105 (4): 993-6, 1999. [PUBMED Abstract]
Martin P, Chadburn A, Christos P, et al.: Intensive treatment strategies may not provide superior outcomes in mantle cell lymphoma: overall survival exceeding 7 years with standard therapies. Ann Oncol 19 (7): 1327-30, 2008. [PUBMED Abstract]
Kyriakou C, Canals C, Cornelissen JJ, et al.: Allogeneic stem-cell transplantation in patients with Waldenström macroglobulinemia: report from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 28 (33): 4926-34, 2010. [PUBMED Abstract]
Leleu X, Soumerai J, Roccaro A, et al.: Increased incidence of transformation and myelodysplasia/acute leukemia in patients with Waldenström macroglobulinemia treated with nucleoside analogs. J Clin Oncol 27 (2): 250-5, 2009. [PUBMED Abstract]
Leblond V, Lévy V, Maloisel F, et al.: Multicenter, randomized comparative trial of fludarabine and the combination of cyclophosphamide-doxorubicin-prednisone in 92 patients with Waldenström macroglobulinemia in first relapse or with primary refractory disease. Blood 98 (9): 2640-4, 2001. [PUBMED Abstract]
Zucca E, Bertoni F: The spectrum of MALT lymphoma at different sites: biological and therapeutic relevance. Blood 127 (17): 2082-92, 2016. [PUBMED Abstract]
Rossi D, Bertoni F, Zucca E: Marginal-Zone Lymphomas. N Engl J Med 386 (6): 568-581, 2022. [PUBMED Abstract]
Thieblemont C, Cascione L, Conconi A, et al.: A MALT lymphoma prognostic index. Blood 130 (12): 1409-1417, 2017. [PUBMED Abstract]
Alderuccio JP, Zhao W, Desai A, et al.: Risk Factors for Transformation to Higher-Grade Lymphoma and Its Impact on Survival in a Large Cohort of Patients With Marginal Zone Lymphoma From a Single Institution. J Clin Oncol : JCO1800138, 2018. [PUBMED Abstract]
Zullo A, Hassan C, Andriani A, et al.: Eradication therapy for Helicobacter pylori in patients with gastric MALT lymphoma: a pooled data analysis. Am J Gastroenterol 104 (8): 1932-7; quiz 1938, 2009. [PUBMED Abstract]
Nakamura S, Sugiyama T, Matsumoto T, et al.: Long-term clinical outcome of gastric MALT lymphoma after eradication of Helicobacter pylori: a multicentre cohort follow-up study of 420 patients in Japan. Gut 61 (4): 507-13, 2012. [PUBMED Abstract]
Wündisch T, Thiede C, Morgner A, et al.: Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 23 (31): 8018-24, 2005. [PUBMED Abstract]
Ye H, Liu H, Raderer M, et al.: High incidence of t(11;18)(q21;q21) in Helicobacter pylori-negative gastric MALT lymphoma. Blood 101 (7): 2547-50, 2003. [PUBMED Abstract]
Lévy M, Copie-Bergman C, Gameiro C, et al.: Prognostic value of translocation t(11;18) in tumoral response of low-grade gastric lymphoma of mucosa-associated lymphoid tissue type to oral chemotherapy. J Clin Oncol 23 (22): 5061-6, 2005. [PUBMED Abstract]
Nakamura S, Ye H, Bacon CM, et al.: Clinical impact of genetic aberrations in gastric MALT lymphoma: a comprehensive analysis using interphase fluorescence in situ hybridisation. Gut 56 (10): 1358-63, 2007. [PUBMED Abstract]
Schechter NR, Yahalom J: Low-grade MALT lymphoma of the stomach: a review of treatment options. Int J Radiat Oncol Biol Phys 46 (5): 1093-103, 2000. [PUBMED Abstract]
Tsang RW, Gospodarowicz MK, Pintilie M, et al.: Stage I and II MALT lymphoma: results of treatment with radiotherapy. Int J Radiat Oncol Biol Phys 50 (5): 1258-64, 2001. [PUBMED Abstract]
Tsang RW, Gospodarowicz MK, Pintilie M, et al.: Localized mucosa-associated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol 21 (22): 4157-64, 2003. [PUBMED Abstract]
Tsai HK, Li S, Ng AK, et al.: Role of radiation therapy in the treatment of stage I/II mucosa-associated lymphoid tissue lymphoma. Ann Oncol 18 (4): 672-8, 2007. [PUBMED Abstract]
De Leo AN, Bates JE, Lockney NA, et al.: Radiotherapy in Early-stage Gastric MALT: Improved Survival Without Increased Cardiac Death. Am J Clin Oncol 43 (11): 770-775, 2020. [PUBMED Abstract]
Gunther JR, Xu J, Bhutani MS, et al.: Response-adapted ultra-low-dose 4 Gy radiation as definitive therapy of gastric MALT lymphoma: a single-centre, pilot trial. Lancet Haematol 11 (7): e521-e529, 2024. [PUBMED Abstract]
Martinelli G, Laszlo D, Ferreri AJ, et al.: Clinical activity of rituximab in gastric marginal zone non-Hodgkin’s lymphoma resistant to or not eligible for anti-Helicobacter pylori therapy. J Clin Oncol 23 (9): 1979-83, 2005. [PUBMED Abstract]
Cogliatti SB, Schmid U, Schumacher U, et al.: Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 101 (5): 1159-70, 1991. [PUBMED Abstract]
Zinzani PL, Magagnoli M, Galieni P, et al.: Nongastrointestinal low-grade mucosa-associated lymphoid tissue lymphoma: analysis of 75 patients. J Clin Oncol 17 (4): 1254, 1999. [PUBMED Abstract]
Thieblemont C, Bastion Y, Berger F, et al.: Mucosa-associated lymphoid tissue gastrointestinal and nongastrointestinal lymphoma behavior: analysis of 108 patients. J Clin Oncol 15 (4): 1624-30, 1997. [PUBMED Abstract]
Pavlick AC, Gerdes H, Portlock CS: Endoscopic ultrasound in the evaluation of gastric small lymphocytic mucosa-associated lymphoid tumors. J Clin Oncol 15 (5): 1761-6, 1997. [PUBMED Abstract]
Morgner A, Miehlke S, Fischbach W, et al.: Complete remission of primary high-grade B-cell gastric lymphoma after cure of Helicobacter pylori infection. J Clin Oncol 19 (7): 2041-8, 2001. [PUBMED Abstract]
Chen LT, Lin JT, Shyu RY, et al.: Prospective study of Helicobacter pylori eradication therapy in stage I(E) high-grade mucosa-associated lymphoid tissue lymphoma of the stomach. J Clin Oncol 19 (22): 4245-51, 2001. [PUBMED Abstract]
Chen LT, Lin JT, Tai JJ, et al.: Long-term results of anti-Helicobacter pylori therapy in early-stage gastric high-grade transformed MALT lymphoma. J Natl Cancer Inst 97 (18): 1345-53, 2005. [PUBMED Abstract]
Kuo SH, Yeh KH, Wu MS, et al.: Helicobacter pylori eradication therapy is effective in the treatment of early-stage H pylori-positive gastric diffuse large B-cell lymphomas. Blood 119 (21): 4838-44; quiz 5057, 2012. [PUBMED Abstract]
Uno T, Isobe K, Shikama N, et al.: Radiotherapy for extranodal, marginal zone, B-cell lymphoma of mucosa-associated lymphoid tissue originating in the ocular adnexa: a multiinstitutional, retrospective review of 50 patients. Cancer 98 (4): 865-71, 2003. [PUBMED Abstract]
Bayraktar S, Bayraktar UD, Stefanovic A, et al.: Primary ocular adnexal mucosa-associated lymphoid tissue lymphoma (MALT): single institution experience in a large cohort of patients. Br J Haematol 152 (1): 72-80, 2011. [PUBMED Abstract]
Stefanovic A, Lossos IS: Extranodal marginal zone lymphoma of the ocular adnexa. Blood 114 (3): 501-10, 2009. [PUBMED Abstract]
Vazquez A, Khan MN, Sanghvi S, et al.: Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue of the salivary glands: a population-based study from 1994 to 2009. Head Neck 37 (1): 18-22, 2015. [PUBMED Abstract]
Raderer M, Streubel B, Woehrer S, et al.: High relapse rate in patients with MALT lymphoma warrants lifelong follow-up. Clin Cancer Res 11 (9): 3349-52, 2005. [PUBMED Abstract]
Sretenovic M, Colovic M, Jankovic G, et al.: More than a third of non-gastric malt lymphomas are disseminated at diagnosis: a single center survey. Eur J Haematol 82 (5): 373-80, 2009. [PUBMED Abstract]
Raderer M, Wöhrer S, Streubel B, et al.: Assessment of disease dissemination in gastric compared with extragastric mucosa-associated lymphoid tissue lymphoma using extensive staging: a single-center experience. J Clin Oncol 24 (19): 3136-41, 2006. [PUBMED Abstract]
Zucca E, Conconi A, Martinelli G, et al.: Final Results of the IELSG-19 Randomized Trial of Mucosa-Associated Lymphoid Tissue Lymphoma: Improved Event-Free and Progression-Free Survival With Rituximab Plus Chlorambucil Versus Either Chlorambucil or Rituximab Monotherapy. J Clin Oncol 35 (17): 1905-1912, 2017. [PUBMED Abstract]
Kiesewetter B, Raderer M: Antibiotic therapy in nongastrointestinal MALT lymphoma: a review of the literature. Blood 122 (8): 1350-7, 2013. [PUBMED Abstract]
Grünberger B, Hauff W, Lukas J, et al.: ‘Blind’ antibiotic treatment targeting Chlamydia is not effective in patients with MALT lymphoma of the ocular adnexa. Ann Oncol 17 (3): 484-7, 2006. [PUBMED Abstract]
Kuo SH, Chen LT, Yeh KH, et al.: Nuclear expression of BCL10 or nuclear factor kappa B predicts Helicobacter pylori-independent status of early-stage, high-grade gastric mucosa-associated lymphoid tissue lymphomas. J Clin Oncol 22 (17): 3491-7, 2004. [PUBMED Abstract]
Desai A, Joag MG, Lekakis L, et al.: Long-term course of patients with primary ocular adnexal MALT lymphoma: a large single-institution cohort study. Blood 129 (3): 324-332, 2017. [PUBMED Abstract]
Herold M, Hoster E, Janssens A, et al.: Immunochemotherapy and Maintenance With Obinutuzumab or Rituximab in Patients With Previously Untreated Marginal Zone Lymphoma in the Randomized GALLIUM Trial. Hemasphere 6 (3): e699, 2022. [PUBMED Abstract]
Salar A, Domingo-Domenech E, Panizo C, et al.: Long-term results of a phase 2 study of rituximab and bendamustine for mucosa-associated lymphoid tissue lymphoma. Blood 130 (15): 1772-1774, 2017. [PUBMED Abstract]
Alderuccio JP, Arcaini L, Watkins MP, et al.: An international analysis evaluating frontline bendamustine with rituximab in extranodal marginal zone lymphoma. Blood Adv 6 (7): 2035-2044, 2022. [PUBMED Abstract]
Opat S, Tedeschi A, Linton K, et al.: The MAGNOLIA Trial: Zanubrutinib, a Next-Generation Bruton Tyrosine Kinase Inhibitor, Demonstrates Safety and Efficacy in Relapsed/Refractory Marginal Zone Lymphoma. Clin Cancer Res 27 (23): 6323-6332, 2021. [PUBMED Abstract]
Noy A, de Vos S, Coleman M, et al.: Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: long-term follow-up and biomarker analysis. Blood Adv 4 (22): 5773-5784, 2020. [PUBMED Abstract]
Leonard JP, Trneny M, Izutsu K, et al.: AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 37 (14): 1188-1199, 2019. [PUBMED Abstract]
Luminari S, Merli M, Rattotti S, et al.: Early progression as a predictor of survival in marginal zone lymphomas: an analysis from the FIL-NF10 study. Blood 134 (10): 798-801, 2019. [PUBMED Abstract]
Vallisa D, Bernuzzi P, Arcaini L, et al.: Role of anti-hepatitis C virus (HCV) treatment in HCV-related, low-grade, B-cell, non-Hodgkin’s lymphoma: a multicenter Italian experience. J Clin Oncol 23 (3): 468-73, 2005. [PUBMED Abstract]
Merli M, Rattotti S, Spina M, et al.: Direct-Acting Antivirals as Primary Treatment for Hepatitis C Virus-Associated Indolent Non-Hodgkin Lymphomas: The BArT Study of the Fondazione Italiana Linfomi. J Clin Oncol 40 (35): 4060-4070, 2022. [PUBMED Abstract]
Lecuit M, Abachin E, Martin A, et al.: Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med 350 (3): 239-48, 2004. [PUBMED Abstract]
Arcaini L, Paulli M, Boveri E, et al.: Splenic and nodal marginal zone lymphomas are indolent disorders at high hepatitis C virus seroprevalence with distinct presenting features but similar morphologic and phenotypic profiles. Cancer 100 (1): 107-15, 2004. [PUBMED Abstract]
Arcaini L, Rossi D, Paulli M: Splenic marginal zone lymphoma: from genetics to management. Blood 127 (17): 2072-81, 2016. [PUBMED Abstract]
Parry-Jones N, Matutes E, Gruszka-Westwood AM, et al.: Prognostic features of splenic lymphoma with villous lymphocytes: a report on 129 patients. Br J Haematol 120 (5): 759-64, 2003. [PUBMED Abstract]
Arcaini L, Lazzarino M, Colombo N, et al.: Splenic marginal zone lymphoma: a prognostic model for clinical use. Blood 107 (12): 4643-9, 2006. [PUBMED Abstract]
Iannitto E, Ambrosetti A, Ammatuna E, et al.: Splenic marginal zone lymphoma with or without villous lymphocytes. Hematologic findings and outcomes in a series of 57 patients. Cancer 101 (9): 2050-7, 2004. [PUBMED Abstract]
Hermine O, Lefrère F, Bronowicki JP, et al.: Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis C virus infection. N Engl J Med 347 (2): 89-94, 2002. [PUBMED Abstract]
Kelaidi C, Rollot F, Park S, et al.: Response to antiviral treatment in hepatitis C virus-associated marginal zone lymphomas. Leukemia 18 (10): 1711-6, 2004. [PUBMED Abstract]
Treatment Option Overview for Indolent B-Cell Non-Hodgkin Lymphoma
Treatment of indolent non-Hodgkin lymphoma (NHL) depends on the histological type and stage. Many of the improvements in survival have been made because of clinical trials that have attempted to improve on conventional or standard therapy.
In asymptomatic patients with indolent forms of advanced NHL, treatment may be deferred until the patient becomes symptomatic as the disease progresses. When treatment is deferred, the clinical course of patients with indolent NHL varies; frequent and careful observation is required so that effective treatment can be initiated when the clinical course of the disease accelerates. Some patients have a prolonged indolent course, but others have disease that rapidly evolves into more aggressive types of NHL that require immediate treatment.
Radiation techniques differ somewhat from those used in the treatment of Hodgkin lymphoma. The dose of radiation therapy usually varies from 25 Gy to 50 Gy and is dependent on factors that include the histological type of lymphoma, the patient’s stage and overall condition, the goal of treatment (curative or palliative), the proximity of sensitive surrounding organs, and whether the patient is being treated with radiation therapy alone or in combination with chemotherapy. Given the patterns of disease presentations and relapse, treatment may need to include unusual sites such as Waldeyer ring, epitrochlear nodes, or mesenteric nodes. The associated morbidity of the treatment must be considered carefully. Most patients who receive radiation are treated on only one side of the diaphragm. Localized presentations of extranodal NHL may be treated with involved-field techniques with significant (>50%) success.
In situations where mediastinal radiation therapy would encompass the left side of the heart or would increase breast cancer risk in young female patients, proton therapy may be considered to reduce radiation dose to organs at risk.[1] For more information, see the Superior Vena Cava Syndrome section in Cardiopulmonary Syndromes.
Table 4. Treatment Options for Indolent B-Cell Non-Hodgkin Lymphoma (NHL)
Stage
Treatment Options
CAR = chimeric antigen receptor; SCT = stem cell transplant.
Indolent Stage I and Indolent, Contiguous Stage II B-cell NHL
Even though existing treatments cure a significant fraction of patients with lymphoma, numerous clinical trials that explore treatment improvements are in progress. If possible, patients can be included in these studies. Standardized guidelines for response assessment have been suggested for use in clinical trials.[2]
Several retrospective reviews suggest that routine surveillance scans offer little to no value in patients with diffuse-large B-cell lymphoma (DLBCL) who have attained a clinical complete remission after induction therapy. Prognostic value is also difficult to identify for an interim positron emission tomography–computed tomography scan during induction therapy for DLBCL.[3–6]
Active hepatitis B or hepatitis C can be assessed before treatment with rituximab and/or chemotherapy.[7,8] Patients with detectable hepatitis B virus (HBV) benefit from prophylaxis with entecavir in the context of rituximab therapy.[9,10] Patients with a resolved HBV infection (defined as hepatitis B surface antigen-negative but hepatitis B core antibody-positive) are at risk of reactivation of HBV and require monitoring of HBV DNA. In a retrospective study of 326 patients, prophylactic nucleoside analogue therapy lowered HBV reactivation from 10.8% to 2.1%.[11] Similarly, prophylaxis for herpes zoster with acyclovir or valacyclovir and prophylaxis for Pneumocystis with trimethoprim/sulfamethoxazole or dapsone are usually given to patients receiving rituximab with or without combination chemotherapy. Long-term impaired immune health was evaluated in a retrospective cohort study of 21,690 survivors of DLBCL from the California Cancer Registry. Elevated incidence rate ratios were found up to 10 years later for pneumonia (10.8-fold), meningitis (5.3-fold), immunoglobulin deficiency (17.6-fold), and autoimmune cytopenias (12-fold).[12]
Among 2,508 patients in a Danish registry, the incidence of doxorubicin-induced congestive heart failure increased for 115 NHL survivors with a history of cardiac disease (hazard ratio [HR], 2.71; 95% confidence interval [CI], 1.15−6.36) and/or multiple cardiovascular risk factors (HR, 2.86; 95% CI, 1.56−5.23).[13]
References
Dabaja BS, Hoppe BS, Plastaras JP, et al.: Proton therapy for adults with mediastinal lymphomas: the International Lymphoma Radiation Oncology Group guidelines. Blood 132 (16): 1635-1646, 2018. [PUBMED Abstract]
Cheson BD, Horning SJ, Coiffier B, et al.: Report of an international workshop to standardize response criteria for non-Hodgkin’s lymphomas. NCI Sponsored International Working Group. J Clin Oncol 17 (4): 1244, 1999. [PUBMED Abstract]
Mamot C, Klingbiel D, Hitz F, et al.: Final Results of a Prospective Evaluation of the Predictive Value of Interim Positron Emission Tomography in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP-14 (SAKK 38/07). J Clin Oncol 33 (23): 2523-9, 2015. [PUBMED Abstract]
Thompson CA, Ghesquieres H, Maurer MJ, et al.: Utility of routine post-therapy surveillance imaging in diffuse large B-cell lymphoma. J Clin Oncol 32 (31): 3506-12, 2014. [PUBMED Abstract]
El-Galaly TC, Jakobsen LH, Hutchings M, et al.: Routine Imaging for Diffuse Large B-Cell Lymphoma in First Complete Remission Does Not Improve Post-Treatment Survival: A Danish-Swedish Population-Based Study. J Clin Oncol 33 (34): 3993-8, 2015. [PUBMED Abstract]
Huntington SF, Svoboda J, Doshi JA: Cost-effectiveness analysis of routine surveillance imaging of patients with diffuse large B-cell lymphoma in first remission. J Clin Oncol 33 (13): 1467-74, 2015. [PUBMED Abstract]
Niitsu N, Hagiwara Y, Tanae K, et al.: Prospective analysis of hepatitis B virus reactivation in patients with diffuse large B-cell lymphoma after rituximab combination chemotherapy. J Clin Oncol 28 (34): 5097-100, 2010. [PUBMED Abstract]
Dong HJ, Ni LN, Sheng GF, et al.: Risk of hepatitis B virus (HBV) reactivation in non-Hodgkin lymphoma patients receiving rituximab-chemotherapy: a meta-analysis. J Clin Virol 57 (3): 209-14, 2013. [PUBMED Abstract]
Huang YH, Hsiao LT, Hong YC, et al.: Randomized controlled trial of entecavir prophylaxis for rituximab-associated hepatitis B virus reactivation in patients with lymphoma and resolved hepatitis B. J Clin Oncol 31 (22): 2765-72, 2013. [PUBMED Abstract]
Li H, Zhang HM, Chen LF, et al.: Prophylactic lamivudine to improve the outcome of HBsAg-positive lymphoma patients during chemotherapy: a systematic review and meta-analysis. Clin Res Hepatol Gastroenterol 39 (1): 80-92, 2015. [PUBMED Abstract]
Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019. [PUBMED Abstract]
Shree T, Li Q, Glaser SL, et al.: Impaired Immune Health in Survivors of Diffuse Large B-Cell Lymphoma. J Clin Oncol 38 (15): 1664-1675, 2020. [PUBMED Abstract]
Salz T, Zabor EC, de Nully Brown P, et al.: Preexisting Cardiovascular Risk and Subsequent Heart Failure Among Non-Hodgkin Lymphoma Survivors. J Clin Oncol 35 (34): 3837-3843, 2017. [PUBMED Abstract]
Treatment of Indolent Stage I and Indolent, Contiguous Stage II B-Cell Non-Hodgkin Lymphoma
Although localized presentations are uncommon in B-cell non-Hodgkin lymphoma (NHL), the goal of treatment is to cure the disease in patients with confirmed localized occurrence after undergoing appropriate staging.
Treatment Options for Indolent Stage I and Indolent, Contiguous Stage II B-Cell NHL
Treatment options for indolent stage I and indolent, contiguous stage II B-cell NHL include:
In a prospective randomized trial, 150 patients with stage I or stage II follicular lymphoma were randomly assigned to 30 Gy of involved-field radiation therapy alone or radiation therapy plus six cycles of R-CVP (rituximab, cyclophosphamide, vincristine, prednisolone). With a median follow-up of 9.6 years, the 10-year progression-free survival (PFS) rate favored combined-modality therapy, at 59% (95% confidence interval [CI], 46%–74%) versus 41% for radiation therapy alone (95% CI, 30%–57%) (P = .033). There was no difference in overall survival (OS) (87% and 95%; P = .40).[1][Level of evidence B1]
The National Lymphocare Study identified 471 patients with stage I follicular lymphoma. Of those patients, 206 were rigorously staged with a bone marrow aspirate and biopsy, and computed tomography (CT) scans or positron emission tomography (PET)-CT scans.[2] Nonrandomized treatments included radiation therapy (27%), rituximab-chemotherapy (R-chemotherapy) (28%), watchful waiting (17%), R-chemotherapy plus radiation therapy (13%), and rituximab alone (12%), although more than one-third of the patients started with watchful waiting. With a median follow-up of 57 months, PFS favored R-chemotherapy or R-chemotherapy plus radiation therapy, but OS was nearly identical, all over 90%.[2][Level of evidence C2] Clinical trials are required to answer the following questions:[3]
If the PET-CT scan is clear after excisional biopsy, is watchful waiting or radiation therapy preferred?
Should rituximab be added to radiation therapy for stage I follicular lymphoma?
Is there any role for R-chemotherapy plus radiation therapy?
Radiation therapy
Long-term disease control within radiation fields can be achieved in a significant number of patients with indolent stage I or stage II NHL by using radiation therapy. This requires dosages of radiation that usually range from 25 Gy to 40 Gy to involved sites or to extended fields that cover adjacent nodal sites.[1,4–6] Almost one-half of all patients treated with radiation therapy alone will have out-of-field relapse within 10 years.[1,6,7]
A retrospective review of 512 patients from an international consortium evaluated patients with early-stage follicular lymphoma who received at least 24 Gy of localized radiation therapy at initial presentation. With a median follow-up of 52 months, 29.1% of patients developed recurrent lymphoma at a median of 23 months (range, 1−143 months).[8] With a median follow-up of 33 months after relapse, the 3-year OS rate was 91.4% after patients received subsequent systemic chemotherapy that involved rituximab.[8]
Very low-dose radiation therapy with 4 Gy (2 Gy × 2 fractions) can result in 50% remission rates for patients who cannot tolerate higher doses.[9] In a multicenter, randomized, prospective trial, 548 patients with follicular or marginal zone lymphoma received radiation therapy, either 4 Gy in 2 fractions or 24 Gy in 12 fractions.[10]
At a median follow-up of 73.8 months, the 5-year local complete response rate was 89.9% (85.5%–93.1%) after 24 Gy and 70.4% (64.7%–75.4%) after 4 Gy (hazard ratio, 3.46; 95% CI, 2.25–5.33; P < .0001).[10]
Although durable local control was superior for patients who received 24 Gy, the 4 Gy regimen was nearly comparable with reductions in radiation exposure, time undergoing therapy, and cost.
Rituximab with or without chemotherapy
When radiation therapy is contraindicated, or when an alternative treatment is preferred, patients with symptomatic disease who require therapy may receive rituximab with or without chemotherapy (as outlined below for more advanced-stage patients).[11] The value of adjuvant treatment with radiation to decrease relapse, plus rituximab (an anti–CD20 monoclonal antibody) either alone or in combination with chemotherapy, has been extrapolated from trials of patients with advanced-stage disease and has not been confirmed.[12,13]
Watchful waiting
Watchful waiting can be considered for asymptomatic patients.[14] Watchful waiting has never been compared with up-front radiation therapy in a prospective randomized trial. A retrospective analysis of the Surveillance, Epidemiology and End Results (SEER) Program database in patients diagnosed over a span of 30 years showed improved outcomes for up-front radiation therapy.[15]
Other therapies as designated for patients with advanced-stage disease
Patients with disease unable to be encompassed by radiation therapy are treated as outlined for patients with stage III or stage IV low-grade lymphoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
MacManus M, Fisher R, Roos D, et al.: Randomized Trial of Systemic Therapy After Involved-Field Radiotherapy in Patients With Early-Stage Follicular Lymphoma: TROG 99.03. J Clin Oncol 36 (29): 2918-2925, 2018. [PUBMED Abstract]
Friedberg JW, Byrtek M, Link BK, et al.: Effectiveness of first-line management strategies for stage I follicular lymphoma: analysis of the National LymphoCare Study. J Clin Oncol 30 (27): 3368-75, 2012. [PUBMED Abstract]
Montoto S: Management of localized-stage follicular lymphoma: changing the paradigm? J Clin Oncol 30 (27): 3328-9, 2012. [PUBMED Abstract]
Haas RL, Poortmans P, de Jong D, et al.: High response rates and lasting remissions after low-dose involved field radiotherapy in indolent lymphomas. J Clin Oncol 21 (13): 2474-80, 2003. [PUBMED Abstract]
Guckenberger M, Alexandrow N, Flentje M: Radiotherapy alone for stage I-III low grade follicular lymphoma: long-term outcome and comparison of extended field and total nodal irradiation. Radiat Oncol 7: 103, 2012. [PUBMED Abstract]
Brady JL, Binkley MS, Hajj C, et al.: Definitive radiotherapy for localized follicular lymphoma staged by 18F-FDG PET-CT: a collaborative study by ILROG. Blood 133 (3): 237-245, 2019. [PUBMED Abstract]
Guadagnolo BA, Li S, Neuberg D, et al.: Long-term outcome and mortality trends in early-stage, Grade 1-2 follicular lymphoma treated with radiation therapy. Int J Radiat Oncol Biol Phys 64 (3): 928-34, 2006. [PUBMED Abstract]
Binkley MS, Brady JL, Hajj C, et al.: Salvage Treatment and Survival for Relapsed Follicular Lymphoma Following Primary Radiation Therapy: A Collaborative Study on Behalf of ILROG. Int J Radiat Oncol Biol Phys 104 (3): 522-529, 2019. [PUBMED Abstract]
Hoskin PJ, Kirkwood AA, Popova B, et al.: 4 Gy versus 24 Gy radiotherapy for patients with indolent lymphoma (FORT): a randomised phase 3 non-inferiority trial. Lancet Oncol 15 (4): 457-63, 2014. [PUBMED Abstract]
Hoskin P, Popova B, Schofield O, et al.: 4 Gy versus 24 Gy radiotherapy for follicular and marginal zone lymphoma (FoRT): long-term follow-up of a multicentre, randomised, phase 3, non-inferiority trial. Lancet Oncol 22 (3): 332-340, 2021. [PUBMED Abstract]
Cartron G, Bachy E, Tilly H, et al.: Randomized Phase III Trial Evaluating Subcutaneous Rituximab for the First-Line Treatment of Low-Tumor Burden Follicular Lymphoma: Results of a LYSA Study. J Clin Oncol 41 (19): 3523-3533, 2023. [PUBMED Abstract]
Kelsey SM, Newland AC, Hudson GV, et al.: A British National Lymphoma Investigation randomised trial of single agent chlorambucil plus radiotherapy versus radiotherapy alone in low grade, localised non-Hodgkins lymphoma. Med Oncol 11 (1): 19-25, 1994. [PUBMED Abstract]
Seymour JF, Pro B, Fuller LM, et al.: Long-term follow-up of a prospective study of combined modality therapy for stage I-II indolent non-Hodgkin’s lymphoma. J Clin Oncol 21 (11): 2115-22, 2003. [PUBMED Abstract]
Advani R, Rosenberg SA, Horning SJ: Stage I and II follicular non-Hodgkin’s lymphoma: long-term follow-up of no initial therapy. J Clin Oncol 22 (8): 1454-9, 2004. [PUBMED Abstract]
Pugh TJ, Ballonoff A, Newman F, et al.: Improved survival in patients with early stage low-grade follicular lymphoma treated with radiation: a Surveillance, Epidemiology, and End Results database analysis. Cancer 116 (16): 3843-51, 2010. [PUBMED Abstract]
Treatment of Indolent, Noncontiguous Stage II/III/IV B-Cell Non-Hodgkin Lymphoma
Optimal treatment of advanced stages of low-grade non-Hodgkin lymphoma (NHL) is controversial because current therapeutic options result in low cure rates. Numerous clinical trials are in progress to evaluate treatment issues, and patients are urged to participate. The rate of relapse is fairly constant over time, even in patients who have achieved complete response to treatment. Relapse may occur many years after treatment. Currently, no randomized trials provide guidance to clinicians about the initial choice of watchful waiting, rituximab, nucleoside analogues, alkylating agents, combination chemotherapy, radiolabeled monoclonal antibodies, or combinations of these options.[1]; [2][Level of evidence B1]
For patients with indolent, noncontiguous stage II and stage III NHL, central lymphatic radiation therapy has been proposed but is not usually recommended as a form of treatment.[3,4]
Patients with a resolved hepatitis B virus (HBV) infection (defined as hepatitis B surface antigen-negative but hepatitis B core antibody-positive) are at risk of reactivation of HBV and require monitoring of HBV DNA. In a retrospective study of 326 patients, prophylactic nucleoside analogue therapy lowered HBV reactivation from 10.8% to 2.1%.[5]
Treatment Options for Indolent, Noncontiguous Stage II/III/IV B-Cell NHL
Treatment options for indolent, noncontiguous stage II/III/IV B-cell NHL include:
Because none of the therapies listed above are curative for advanced-stage disease, innovative approaches are under clinical evaluation.
Watchful waiting for asymptomatic patients
Because relapse may occur many years after treatment, even in patients who have achieved complete responses, deferred treatment (i.e., watchful waiting until the patient becomes symptomatic before initiating treatment) can be considered.[2,6–8] The Follicular Lymphoma International Prognostic Index (FLIPI) and the revised FLIPI-2 can predict progression-free survival (PFS) and overall survival (OS), but the scores cannot be used to establish the need for therapy in asymptomatic patients.[9,10]
Evidence (watchful waiting):
Three randomized trials compared watchful waiting with immediate chemotherapy.[7,11]; [12][Level of evidence A1]
All three trials showed no difference in cause-specific survival or OS.
For patients randomly assigned to watchful waiting, the median time to require therapy was 2 to 3 years. One-third of patients undergoing watchful waiting never required treatment (one-half died of other causes and the other half remained progression free after 10 years).
A selected group of 107 patients with advanced-stage follicular lymphoma were managed with initial watchful waiting; subsequent therapy was delayed for a median of 55 months. These patients achieved equivalent freedom from treatment failure and OS compared with a similar cohort treated immediately with rituximab.[13][Level of evidence C2] This implies that watchful waiting remains a relevant approach even in the rituximab era.
Rituximab alone or in combination with cytotoxic agents used in front-line therapy
Standard therapy includes rituximab, an anti–CD20 monoclonal antibody, either alone, as was shown in the ECOG-E4402 trial (NCT00075946),[14–19] or in combination with purine nucleoside analogues, such as fludarabine or cladribine, alkylating agents (with or without steroids), or combination chemotherapy. Rituximab may be considered as first-line therapy, either alone or in combination with other agents. Rituximab may be given intravenously (IV) or subcutaneously (SQ), and biosimilar versions, such as CT-P10 and GP2013, have shown equivalent efficacy and safety.[20–22] Combinations include the following:
R-CHOP: rituximab + cyclophosphamide + doxorubicin + vincristine + prednisone.[29–33] A Cochrane meta-analysis could not identify any OS benefit of adding doxorubicin to chemotherapy regimens with rituximab or to chemotherapy regimens without rituximab.[34][Level of evidence A1]
Evidence (rituximab with or without chemotherapy):
A prospective randomized trial of 534 patients with previously untreated, advanced-stage, follicular lymphoma compared R-CHOP, R-FM, and R-CVP.[29]
With a median follow-up of 84 months, there was no difference in OS (8-year OS rate, 83%; 95% confidence interval [CI], 79%–87%), but the 8-year PFS rates favored R-CHOP (52%) and R-FM (49%) over R-CVP (42%) (P for the three regimens = .037).[29][Level of evidence B1]
Four randomized prospective studies of previously untreated patients (involving more than 1,300 patients) and one Cochrane meta-analysis that included both untreated and previously treated patients (involving almost 1,000 patients) have compared rituximab plus combination chemotherapy with chemotherapy alone.[28,33,37]; [38][Level of evidence A1]
Rituximab plus chemotherapy was superior in terms of event-free survival (EFS) or PFS (ranging from 2–3 years) in all of the studies and in terms of OS in all but one study (absolute benefit ranging from 6%–13% at 4 years, P < .04; hazard ratio [HR], 0.63 [0.51–0.79] for the meta-analysis).
All of these trials were performed in symptomatic patients who required therapy. These results do not negate watchful waiting when appropriate.
Fluorine F 18-fludeoxyglucose positron emission tomography–computed tomography (18F-FDG PET-CT) scan status at the completion of rituximab plus chemotherapy induction therapy is strongly predictive of outcome. It is not yet known whether acting on the results of the scans translates into better outcomes.[39,40]
In a prospective randomized trial (NCT00991211), 527 patients with indolent and mantle cell lymphoma were randomly assigned to receive either bendamustine and rituximab or R-CHOP.[24][Level of evidence B1]
With a median follow-up of 45 months, the median PFS favored the bendamustine arm (69 months vs. 31 months [HR, 0.58; 95% CI, 0.44–0.74; P < .0001]) but with no difference in OS.
Compared with the R-CHOP arm, the bendamustine arm was associated with significantly lower rates of alopecia, hematologic toxicity, stomatitis, peripheral neuropathy, and infections.
In a similar prospective randomized trial, 447 patients with indolent and mantle cell lymphoma were assigned to bendamustine and rituximab versus R-CHOP or R-CVP.[25][Level of evidence B1]
With a median follow-up of 65 months, the 5-year PFS rate favored bendamustine and rituximab, 65.5% versus 55.8% (HR, 0.61; 95% CI, 0.45–0.85; P = .0025), but with no difference in OS.
Increased deaths in the bendamustine-and-rituximab arm from cardiovascular causes (seven vs. one) and from secondary malignancies other than lymphoma (five vs. three) may have contributed to the lack of OS advantage.
Lenalidomide and rituximab
The combination of the immunomodulating agent lenalidomide with rituximab (the so-called R2 regimen) has been proposed as an alternative regimen to combinations involving cytotoxic agents and their subsequent short- and long-term toxicities.
Evidence (lenalidomide and rituximab):
In a randomized prospective trial (RELEVANCE [NCT01650701]) of 1,030 patients with previously untreated follicular lymphoma, rituximab plus lenalidomide for 18 months was compared with rituximab plus chemotherapy (usually R-CHOP).[41,42] All patients received rituximab maintenance for up to 2 years.
With a median follow-up of 72 months, the 6-year PFS rates (60% and 59%) and 3-year OS rates (89%) were identical (HR for PFS, 1.03; 95% CI, 0.84–1.27; P = .78) (HR for OS was not reported).[41,42][Level of evidence A1]
This trial established that the R2 regimen is as effective as rituximab plus cytotoxic chemotherapy options. The transformation rate to aggressive lymphoma per year was 0.68% in the R2 group and 0.45% in the R-chemotherapy group. With a median follow-up of 72 months, there were no new safety signals.[42]
In a randomized prospective trial of 358 patients with resistant/refractory indolent lymphoma (usually follicular lymphoma), the R2 regimen was compared with rituximab alone.[43]
With a median follow-up of 28 months, the median PFS was 39.4 months for R2 and 14.1 months for rituximab alone (P < .0001), with no difference in OS.[43][Level of evidence B1]
Maintenance rituximab
After induction therapy with rituximab only or with rituximab plus chemotherapy, rituximab can be used once every 2 to 3 months as maintenance therapy. Several studies have evaluated this approach.
Evidence (maintenance rituximab for previously untreated patients):
In the PRIMA study (NCT00140582), 1,018 patients with high-risk, previously untreated, symptomatic disease achieved complete response or partial response after induction therapy with immunochemotherapy (usually R-CHOP). Patients were then randomly assigned to 2 years of maintenance rituximab versus no maintenance.[44][Level of evidence B1]
With a median follow-up of 9.0 years, median PFS favored rituximab maintenance (10.5 years) compared with observation (4.1 years) (HR, 0.61; 95% CI, 0.52−0.73; P < .001), but with no difference in OS.
In the United Kingdom/International Study (NCT00112931), 379 previously untreated patients with asymptomatic, low-burden disease were randomly assigned to watchful waiting versus rituximab induction only versus rituximab induction followed by 2 years of rituximab maintenance.[45][Level of evidence A3]
Although OS and histological transformation rates were not different at 3 years, maintenance rituximab was favored based on quality-of-life studies (Mental Adjustment to Cancer Scale P = .0004 at 7 months; Illness Coping Score P = .0012 at 7 months) and time-to-initiation of new treatment by 3 years (54% for watchful waiting vs. 12% for rituximab maintenance [HR, 0.21; 95% CI, 0.14–0.31; P < .0001]).[45][Level of evidence A3]
This study suggested that for some patients, watch and wait resulted in watch and worry.[46] However, from the perspective of OS and histological transformation rates, no benefit could be seen with rituximab maintenance.
In the RESORT study (NCT00075946), 289 previously untreated patients with asymptomatic, low-burden disease were randomly assigned to receive rituximab induction alone, with a re-treatment strategy that used rituximab at relapse versus rituximab induction plus maintenance rituximab every 13 weeks until treatment failure.[47][Level of evidence B3]
With a median follow-up of 8.7 years, the 7-year rate for freedom from cytotoxic chemotherapy or radiation therapy was 83% for patients who received maintenance rituximab and 63% for patients who received induction alone (HR, 2.37; 95% CI, 1.5–3.8; P = .0001). There was no difference in the 10-year OS rate (83% vs. 84%; nonsignificant P = .5972). A re-treatment strategy achieved comparable disease control using significantly fewer doses of rituximab.
Maintenance rituximab induces prolonged B-cell depletion, but morbidity from infections was not evaluated after 2015, when the full protocol data stopped (12 years after the study began).
These three randomized trials in previously untreated patients showed no advantage for the use of rituximab maintenance versus observation and reinduction of therapy at the time of relapse. The trials suggest a benefit for maintenance rituximab after reinduction for relapsed disease. Many questions remain about rituximab maintenance, particularly about truncating therapy at 2 years and long-term safety and efficacy. A trial extending rituximab maintenance to 5 years showed similar EFS or OS versus 1 year of maintenance after induction therapy with rituximab in previously untreated patients.[48][Level of evidence A1]
The FOLL12 study (NCT02063685) included 807 patients with previously untreated high-tumor burden follicular lymphoma. Patients received rituximab plus chemotherapy induction and were randomly assigned to either standard rituximab maintenance (every 8 weeks for 2 years) or to postinduction treatment (monitoring, rituximab maintenance, or radioimmunotherapy) based on their complete metabolic response and measurable residual disease (MRD)–negative status.[49]
With a median follow-up of 53 months, the 3-year PFS rate was 86% for patients who received standard maintenance and 72% for patients who received response-based treatment (P < .001). The 3-year OS rate was the same in both groups (98% vs. 97%; P = .238).[49][Level of evidence B1]
This trial does not support the use of an end-of-treatment PET-CT scan to guide the use of maintenance rituximab.
A prospective trial included 202 patients with previously untreated low-tumor burden follicular lymphoma. Patients were randomly assigned to receive either four weekly doses of IV rituximab (standard dose, 375 mg/m2) or one dose of IV rituximab followed by three weekly doses of SQ rituximab (1,400 mg) and maintenance doses in months 3, 5, 7, and 9.[19]
With a median follow-up of 50.2 months, the 4-year PFS rate was 58.1% (95% CI, 47.5%–67.4%) for patients in the SQ arm who received maintenance therapy, and 41.2% (95% CI, 30.6%–51.6%) (HR, 0.585; 0.939–0.871; P = .0076) for the patients in the IV arm who did not receive maintenance therapy.[19][Level of evidence B1]
There was no difference in OS or time-to-next treatment.
Outside the context of clinical trials, the use of MRD testing has not been shown to be predictive in directing therapy for patients with follicular lymphoma. In retrospective analyses of two randomized prospective trials, while MRD negativity was prognostic of outcome, maintenance rituximab or obinutuzumab prolonged PFS the most among patients with MRD-negative disease.[49,50][Level of evidence C2] Stopping maintenance rituximab or obinutuzumab was not indicated in patients with MRD-negative disease, negating any possible change in therapy based on that status.
In summary, for previously untreated patients, all of the studies showed improved PFS, with no change in OS.
Evidence (maintenance rituximab for previously treated patients):
In a prospective randomized trial of 465 patients with relapsed follicular lymphoma, responders to R-CHOP or CHOP were further randomly assigned to receive rituximab maintenance (1 dose every 3 months for 2 years) or no maintenance.[51][Level of evidence B1]
At a median follow-up of 6 years, rituximab maintenance was better for median PFS (44 months vs. 16 months, P < .001) and borderline for 5-year OS (74% vs. 64%, P = .07).
This benefit for maintenance was evident even for patients who received rituximab during induction therapy. Most patients in both arms received extensive rituximab during postprotocol salvage treatment.
In a prospective randomized trial of 280 patients with relapsed follicular lymphoma, responders to chemotherapy and autologous stem cell transplant consolidation were randomly assigned to receive four doses of rituximab maintenance or no maintenance.[52][Level of evidence B1]
With an 8.3-year median follow-up, the 10-year PFS rates favored maintenance (54% vs. 37% [HR, 0.66; 95% CI, 0.47–0.91; P = .012]), but there was no difference in OS.
A meta-analysis of nine randomized clinical trials with a total of 2,586 patients with follicular lymphoma, most of whom had relapsed disease, compared rituximab maintenance with no maintenance and showed improved OS for rituximab maintenance in previously treated patients (HRdeath, 0.72; 95% CI, 0.57–0.91).[53][Level of evidence A1]
For previously treated patients, there is more evidence to suggest an OS advantage with the use of rituximab maintenance.
Obinutuzumab alone or in combination with cytotoxic agents used in front-line therapy
Obinutuzumab is a glycoengineered type II anti–CD20 monoclonal antibody with greater antibody-dependent cellular cytotoxicity than rituximab.
Evidence (obinutuzumab):
A prospective randomized trial (NCT01332968) of 1,202 patients with previously untreated follicular lymphoma compared obinutuzumab combined with bendamustine (50%), CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) (33%), or CVP (cyclophosphamide, vincristine, and prednisone) (10%) with rituximab combined with the same chemotherapy regimens (based on investigator choice).[54] After six cycles of combination therapy, patients had 2 years of maintenance therapy, receiving the same antibody every 2 months.
With a median follow-up of 34.5 months, the 3-year PFS rate was 80% in the obinutuzumab group and 73.3% in the rituximab group (HR, 0.66; 95% CI, 0.51–0.85; P = .001).[54][Level of evidence B1]
There was no difference in OS.
Compared with what has been seen historically, there was a high rate of toxic deaths among patients using bendamustine in the obinutuzumab arm (5.6%) and in the rituximab arm (4.4%). For patients with indolent low-grade lymphoma, with median survivals exceeding 15 years, the number of toxic deaths during first-line therapy seems excessive. By comparison, the toxic death rate was 1% to 2% when either antibody was combined with CHOP or CVP.
Several issues have been raised about this study:
The side effects were significantly higher with obinutuzumab in terms of infusion reactions and subsequent adverse events.
Obinutuzumab costs significantly more than rituximab.
In summary, in the absence of any change in OS, switching from rituximab to obinutuzumab in combination with chemotherapy for previously untreated follicular lymphoma is a difficult choice. The PFS differences may be attributable to the imbalance in monoclonal antibody dosing, and the increased side effects and costs are mitigating factors. In this trial, bendamustine combined with either antibody led to unacceptable rates of toxic death.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Hagenbeek A, Eghbali H, Monfardini S, et al.: Phase III intergroup study of fludarabine phosphate compared with cyclophosphamide, vincristine, and prednisone chemotherapy in newly diagnosed patients with stage III and IV low-grade malignant Non-Hodgkin’s lymphoma. J Clin Oncol 24 (10): 1590-6, 2006. [PUBMED Abstract]
Gribben JG: How I treat indolent lymphoma. Blood 109 (11): 4617-26, 2007. [PUBMED Abstract]
Jacobs JP, Murray KJ, Schultz CJ, et al.: Central lymphatic irradiation for stage III nodular malignant lymphoma: long-term results. J Clin Oncol 11 (2): 233-8, 1993. [PUBMED Abstract]
Mendenhall NP, Million RR: Comprehensive lymphatic irradiation for stage II-III non-Hodgkin’s lymphoma. Am J Clin Oncol 12 (3): 190-4, 1989. [PUBMED Abstract]
Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019. [PUBMED Abstract]
Ardeshna KM, Smith P, Norton A, et al.: Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a randomised controlled trial. Lancet 362 (9383): 516-22, 2003. [PUBMED Abstract]
Portlock CS, Rosenberg SA: No initial therapy for stage III and IV non-Hodgkin’s lymphomas of favorable histologic types. Ann Intern Med 90 (1): 10-13, 1979.
Solal-Céligny P, Roy P, Colombat P, et al.: Follicular lymphoma international prognostic index. Blood 104 (5): 1258-65, 2004. [PUBMED Abstract]
Federico M, Bellei M, Marcheselli L, et al.: Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol 27 (27): 4555-62, 2009. [PUBMED Abstract]
Brice P, Bastion Y, Lepage E, et al.: Comparison in low-tumor-burden follicular lymphomas between an initial no-treatment policy, prednimustine, or interferon alfa: a randomized study from the Groupe d’Etude des Lymphomes Folliculaires. Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol 15 (3): 1110-7, 1997. [PUBMED Abstract]
Young RC, Longo DL, Glatstein E, et al.: The treatment of indolent lymphomas: watchful waiting v aggressive combined modality treatment. Semin Hematol 25 (2 Suppl 2): 11-6, 1988. [PUBMED Abstract]
Solal-Céligny P, Bellei M, Marcheselli L, et al.: Watchful waiting in low-tumor burden follicular lymphoma in the rituximab era: results of an F2-study database. J Clin Oncol 30 (31): 3848-53, 2012. [PUBMED Abstract]
Ghielmini M, Schmitz SF, Cogliatti SB, et al.: Prolonged treatment with rituximab in patients with follicular lymphoma significantly increases event-free survival and response duration compared with the standard weekly x 4 schedule. Blood 103 (12): 4416-23, 2004. [PUBMED Abstract]
Witzig TE, Vukov AM, Habermann TM, et al.: Rituximab therapy for patients with newly diagnosed, advanced-stage, follicular grade I non-Hodgkin’s lymphoma: a phase II trial in the North Central Cancer Treatment Group. J Clin Oncol 23 (6): 1103-8, 2005. [PUBMED Abstract]
Hainsworth JD, Litchy S, Shaffer DW, et al.: Maximizing therapeutic benefit of rituximab: maintenance therapy versus re-treatment at progression in patients with indolent non-Hodgkin’s lymphoma–a randomized phase II trial of the Minnie Pearl Cancer Research Network. J Clin Oncol 23 (6): 1088-95, 2005. [PUBMED Abstract]
Kahl BS, Hong F, Williams ME, et al.: Rituximab extended schedule or re-treatment trial for low-tumor burden follicular lymphoma: eastern cooperative oncology group protocol e4402. J Clin Oncol 32 (28): 3096-102, 2014. [PUBMED Abstract]
Buske C, Hiddemann W: Rituximab maintenance therapy in indolent NHL: a clinical review. Leuk Res 30 (Suppl 1): S11-5, 2006. [PUBMED Abstract]
Cartron G, Bachy E, Tilly H, et al.: Randomized Phase III Trial Evaluating Subcutaneous Rituximab for the First-Line Treatment of Low-Tumor Burden Follicular Lymphoma: Results of a LYSA Study. J Clin Oncol 41 (19): 3523-3533, 2023. [PUBMED Abstract]
Kim WS, Buske C, Ogura M, et al.: Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol 4 (8): e362-e373, 2017. [PUBMED Abstract]
Davies A, Merli F, Mihaljević B, et al.: Efficacy and safety of subcutaneous rituximab versus intravenous rituximab for first-line treatment of follicular lymphoma (SABRINA): a randomised, open-label, phase 3 trial. Lancet Haematol 4 (6): e272-e282, 2017. [PUBMED Abstract]
Jurczak W, Moreira I, Kanakasetty GB, et al.: Rituximab biosimilar and reference rituximab in patients with previously untreated advanced follicular lymphoma (ASSIST-FL): primary results from a confirmatory phase 3, double-blind, randomised, controlled study. Lancet Haematol 4 (8): e350-e361, 2017. [PUBMED Abstract]
Robinson KS, Williams ME, van der Jagt RH, et al.: Phase II multicenter study of bendamustine plus rituximab in patients with relapsed indolent B-cell and mantle cell non-Hodgkin’s lymphoma. J Clin Oncol 26 (27): 4473-9, 2008. [PUBMED Abstract]
Rummel MJ, Niederle N, Maschmeyer G, et al.: Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381 (9873): 1203-10, 2013. [PUBMED Abstract]
Flinn IW, van der Jagt R, Kahl B, et al.: First-Line Treatment of Patients With Indolent Non-Hodgkin Lymphoma or Mantle-Cell Lymphoma With Bendamustine Plus Rituximab Versus R-CHOP or R-CVP: Results of the BRIGHT 5-Year Follow-Up Study. J Clin Oncol 37 (12): 984-991, 2019. [PUBMED Abstract]
Czuczman MS, Koryzna A, Mohr A, et al.: Rituximab in combination with fludarabine chemotherapy in low-grade or follicular lymphoma. J Clin Oncol 23 (4): 694-704, 2005. [PUBMED Abstract]
Marcus R, Imrie K, Belch A, et al.: CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood 105 (4): 1417-23, 2005. [PUBMED Abstract]
Marcus R, Imrie K, Solal-Celigny P, et al.: Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol 26 (28): 4579-86, 2008. [PUBMED Abstract]
Federico M, Luminari S, Dondi A, et al.: R-CVP versus R-CHOP versus R-FM for the initial treatment of patients with advanced-stage follicular lymphoma: results of the FOLL05 trial conducted by the Fondazione Italiana Linfomi. J Clin Oncol 31 (12): 1506-13, 2013. [PUBMED Abstract]
Luminari S, Ferrari A, Manni M, et al.: Long-Term Results of the FOLL05 Trial Comparing R-CVP Versus R-CHOP Versus R-FM for the Initial Treatment of Patients With Advanced-Stage Symptomatic Follicular Lymphoma. J Clin Oncol 36 (7): 689-696, 2018. [PUBMED Abstract]
Czuczman MS, Weaver R, Alkuzweny B, et al.: Prolonged clinical and molecular remission in patients with low-grade or follicular non-Hodgkin’s lymphoma treated with rituximab plus CHOP chemotherapy: 9-year follow-up. J Clin Oncol 22 (23): 4711-6, 2004. [PUBMED Abstract]
Hainsworth JD, Litchy S, Morrissey LH, et al.: Rituximab plus short-duration chemotherapy as first-line treatment for follicular non-Hodgkin’s lymphoma: a phase II trial of the Minnie Pearl Cancer Research Network. J Clin Oncol 23 (7): 1500-6, 2005. [PUBMED Abstract]
Hiddemann W, Kneba M, Dreyling M, et al.: Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 106 (12): 3725-32, 2005. [PUBMED Abstract]
Itchaki G, Gafter-Gvili A, Lahav M, et al.: Anthracycline-containing regimens for treatment of follicular lymphoma in adults. Cochrane Database Syst Rev 7: CD008909, 2013. [PUBMED Abstract]
Zinzani PL, Pulsoni A, Perrotti A, et al.: Fludarabine plus mitoxantrone with and without rituximab versus CHOP with and without rituximab as front-line treatment for patients with follicular lymphoma. J Clin Oncol 22 (13): 2654-61, 2004. [PUBMED Abstract]
Forstpointner R, Dreyling M, Repp R, et al.: The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 104 (10): 3064-71, 2004. [PUBMED Abstract]
Herold M, Haas A, Srock S, et al.: Rituximab added to first-line mitoxantrone, chlorambucil, and prednisolone chemotherapy followed by interferon maintenance prolongs survival in patients with advanced follicular lymphoma: an East German Study Group Hematology and Oncology Study. J Clin Oncol 25 (15): 1986-92, 2007. [PUBMED Abstract]
Salles GA, Mounier N, de Guibert S, et al.: Rituximab combined with chemotherapy and interferon in follicular lymphoma patients: final analysis of the GELA-GOELAMS FL2000 study with a 5-year follow-up. [Abstract] Blood 110 (11): A-792, 2007.
Dupuis J, Berriolo-Riedinger A, Julian A, et al.: Impact of [(18)F]fluorodeoxyglucose positron emission tomography response evaluation in patients with high-tumor burden follicular lymphoma treated with immunochemotherapy: a prospective study from the Groupe d’Etudes des Lymphomes de l’Adulte and GOELAMS. J Clin Oncol 30 (35): 4317-22, 2012. [PUBMED Abstract]
Trotman J, Fournier M, Lamy T, et al.: Positron emission tomography-computed tomography (PET-CT) after induction therapy is highly predictive of patient outcome in follicular lymphoma: analysis of PET-CT in a subset of PRIMA trial participants. J Clin Oncol 29 (23): 3194-200, 2011. [PUBMED Abstract]
Morschhauser F, Fowler NH, Feugier P, et al.: Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N Engl J Med 379 (10): 934-947, 2018. [PUBMED Abstract]
Morschhauser F, Nastoupil L, Feugier P, et al.: Six-Year Results From RELEVANCE: Lenalidomide Plus Rituximab (R2) Versus Rituximab-Chemotherapy Followed by Rituximab Maintenance in Untreated Advanced Follicular Lymphoma. J Clin Oncol 40 (28): 3239-3245, 2022. [PUBMED Abstract]
Leonard JP, Trneny M, Izutsu K, et al.: AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 37 (14): 1188-1199, 2019. [PUBMED Abstract]
Bachy E, Seymour JF, Feugier P, et al.: Sustained Progression-Free Survival Benefit of Rituximab Maintenance in Patients With Follicular Lymphoma: Long-Term Results of the PRIMA Study. J Clin Oncol 37 (31): 2815-2824, 2019. [PUBMED Abstract]
Ardeshna KM, Qian W, Smith P, et al.: Rituximab versus a watch-and-wait approach in patients with advanced-stage, asymptomatic, non-bulky follicular lymphoma: an open-label randomised phase 3 trial. Lancet Oncol 15 (4): 424-35, 2014. [PUBMED Abstract]
Ansell SM: Follicular lymphoma: watch and wait is watch and worry. Lancet Oncol 15 (4): 368-9, 2014. [PUBMED Abstract]
Kahl BS, Jegede OA, Peterson C, et al.: Long-Term Follow-Up of the RESORT Study (E4402): A Randomized Phase III Comparison of Two Different Rituximab Dosing Strategies for Low-Tumor Burden Follicular Lymphoma. J Clin Oncol 42 (7): 774-778, 2024. [PUBMED Abstract]
Taverna C, Martinelli G, Hitz F, et al.: Rituximab Maintenance for a Maximum of 5 Years After Single-Agent Rituximab Induction in Follicular Lymphoma: Results of the Randomized Controlled Phase III Trial SAKK 35/03. J Clin Oncol 34 (5): 495-500, 2016. [PUBMED Abstract]
Luminari S, Manni M, Galimberti S, et al.: Response-Adapted Postinduction Strategy in Patients With Advanced-Stage Follicular Lymphoma: The FOLL12 Study. J Clin Oncol 40 (7): 729-739, 2022. [PUBMED Abstract]
Pott C, Jurinovic V, Trotman J, et al.: Minimal Residual Disease Status Predicts Outcome in Patients With Previously Untreated Follicular Lymphoma: A Prospective Analysis of the Phase III GALLIUM Study. J Clin Oncol 42 (5): 550-561, 2024. [PUBMED Abstract]
van Oers MH, Tönnissen E, Van Glabbeke M, et al.: BCL-2/IgH polymerase chain reaction status at the end of induction treatment is not predictive for progression-free survival in relapsed/resistant follicular lymphoma: results of a prospective randomized EORTC 20981 phase III intergroup study. J Clin Oncol 28 (13): 2246-52, 2010. [PUBMED Abstract]
Pettengell R, Schmitz N, Gisselbrecht C, et al.: Rituximab purging and/or maintenance in patients undergoing autologous transplantation for relapsed follicular lymphoma: a prospective randomized trial from the lymphoma working party of the European group for blood and marrow transplantation. J Clin Oncol 31 (13): 1624-30, 2013. [PUBMED Abstract]
Vidal L, Gafter-Gvili A, Salles G, et al.: Rituximab maintenance for the treatment of patients with follicular lymphoma: an updated systematic review and meta-analysis of randomized trials. J Natl Cancer Inst 103 (23): 1799-806, 2011. [PUBMED Abstract]
Marcus R, Davies A, Ando K, et al.: Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N Engl J Med 377 (14): 1331-1344, 2017. [PUBMED Abstract]
Treatment of Indolent, Recurrent B-Cell Non-Hodgkin Lymphoma
In general, treatment with standard agents rarely produces a cure in patients with relapsed B-cell non-Hodgkin Lymphoma (NHL). Sustained remissions after relapse can often be obtained in patients with indolent lymphomas, but relapse will usually ensue. Favorable survival after relapse has been associated with an age younger than 60 years, complete remission rather than partial remission, and duration of response longer than 2 years.[1] Even patients in the most favorable subset, however, have a tenfold greater mortality compared with age-adjusted U.S. population rates.[2]
Patients who experience a relapse of indolent lymphoma can often achieve disease control with single-agent or combination chemotherapy, rituximab (an anti–CD20 monoclonal antibody), lenalidomide, radiolabeled anti–CD20 monoclonal antibodies, or palliative radiation therapy.[3,4] However, long-term freedom from second relapse is uncommon and multiple relapses usually occur. Patients with indolent lymphoma may experience a relapse with a more aggressive histology. If the clinical pattern of relapse suggests that the disease is behaving in a more aggressive manner, a biopsy can be performed. If a more aggressive histology is confirmed, therapy must change to a regimen applicable to that histological type.[5] Rapid growth or discordant growth between various disease sites may indicate a histological conversion.
In a retrospective review of 325 patients diagnosed between 1972 and 1999, the 10-year risk of histological transformation was 30%.[6] In this series, high-risk factors for subsequent histological transformation were advanced stage, high-risk Follicular Lymphoma International Prognostic Index, and watchful waiting (as opposed to treatment being initiated at diagnosis). The median survival after transformation was 1 to 2 years, with 25% of patients alive at 5 years and with approximately 10% to 20% of patients alive 10 years after re-treatment.[7]
A prospective trial of 631 patients with follicular lymphoma and with a median follow-up of 60 months in the rituximab era (2002–2009) found a 5-year transformation rate to a higher-grade histology of 11%.[8] The median overall survival (OS) after transformation was 50 months, and the 5-year OS rate was 66%, if the transformation occurred more than 18 months after a diagnosis of follicular lymphoma. This series described a better prognosis for patients with transformation than in the era before rituximab.
For descriptions of the regimens used to treat histological conversions, see the Treatment of Aggressive, Recurrent B-Cell Non-Hodgkin Lymphoma section in Aggressive B-Cell Non-Hodgkin Lymphoma Treatment. The durability of the second remission may be short, and clinical trials can be considered.
Treatment Options for Indolent, Recurrent B-Cell NHL
Treatment options for indolent, recurrent B-cell NHL include:
Rituximab alone or in combination with cytotoxic agents used in front-line therapy
Rituximab results in a 40% to 50% response rate in patients with relapsed indolent B-cell lymphomas.[9–13] Rituximab can also be combined with combination chemotherapy.[14,15]
Evidence (rituximab):
In three randomized, prospective studies involving previously treated patients with relapsed indolent lymphoma, patients were randomly assigned to rituximab maintenance after re-treatment with combination chemotherapy (with or without rituximab during induction) or rituximab alone.[16–18]
All trials showed prolongation of response duration.[16–18] One trial with a median follow-up of 39 months demonstrated improvement in median progression-free survival (PFS) (3.7 years vs. 1.3 years, P < .001) and 5-year OS rate (74% vs. 64%, P = .07), favoring maintenance rituximab.[17]
Obinutuzumab alone or in combination with cytotoxic agents used in front-line therapy
Obinutuzumab is a CD20-binding monoclonal antibody with alternative epitope binding.
Evidence (obinutuzumab):
A randomized prospective trial (NCT01059630) included 396 patients with rituximab-refractory indolent lymphoma (mostly follicular lymphoma). Patients received obinutuzumab plus bendamustine, followed by obinutuzumab maintenance therapy, for 2 years versus bendamustine alone with no maintenance therapy.[19,20][Level of evidence A1]
With a median follow-up of 31.8 months, the 2-year OS rate favored the obinutuzumab combination (74.5% vs. 65.1%) (hazard ratio [HR], 0.67; 95% confidence interval [CI], 0.47–0.96; P = .027). The median PFS also favored the obinutuzumab combination (25.8 months [95% CI, 19.5–41.1] vs. 14.1 months [95% CI, 12.6–16.0]) (HR, 0.57; 95% CI, 0.44–0.73; P < .001).[20][Level of evidence A1]
The contribution of maintenance therapy to the outcome could not be assessed in this design.
Lenalidomide and rituximab
Responses of 20% to 56% have been reported for lenalidomide, especially in patients with follicular lymphoma and small lymphocytic lymphoma, with even higher responses noted for the combination of lenalidomide and rituximab.[21,22][Level of evidence C3]
Zanubrutinib and obinutuzumab
Evidence (zanubrutinib and obinutuzumab):
A randomized, multicenter, phase II study (ROSEWOOD [NCT03332017]) included 217 patients with relapsed or refractory follicular lymphoma after receiving two or more prior lines of therapy. Patients received either the oral Bruton tyrosine kinase inhibitor zanubrutinib plus obinutuzumab or obinutuzumab alone until disease progression.[23][Level of evidence B1]
With a median follow-up of 19.0 months, the median PFS was 28.0 months for patients who received zanubrutinib plus obinutuzumab and 10.4 months for patients who received obinutuzumab alone (HR, 0.50; 95% CI, 0.33–0.75; P < .001).
The overall response rate was 69% (95% CI, 61%–76%) in the zanubrutinib-plus-obinutuzumab arm and 46% (95% CI, 34%–58%) in the obinutuzumab-alone arm (P = .0012).
The U.S. Food and Drug Administration approved zanubrutinib and obinutuzumab for patients with relapsed or refractory follicular lymphoma after two or more prior lines of therapy.
EZH2 inhibitor
Tazemetostat
Tazemetostat is an inhibitor of EZH2, a histone methyltransferase essential to the formation of lymph node germinal centers, especially with activating variants of EZH2.
Evidence (tazemetostat):
A phase II study included 99 patients with relapsed or refractory follicular lymphoma, 45 of whom had activating EZH2 variants, and 54 of whom had wild-type EZH2.[24]
Treatment with tazemetostat resulted in an objective response rate of 69% (95% CI, 53%–82%) for patients with activating EZH2 variants versus 35% (95% CI, 23%–49%) for patients with wild-type EZH2.[24]
With a median follow-up of 22 months, the median PFS was 13.8 months (95% CI, 10.7–22.0) for patients with activating EZH2 variants and 11.1 months (95% CI, 3.7–14.6) for patients with wild-type EZH2.[24][Level of evidence C3]
Grade 3 or 4 treatment-related adverse events occurred in 4% of patients.
Bispecific T-cell engagers
Bispecific T-cell engagers bind to CD20 (or CD19) and to CD3 to direct T cells to eliminate malignant B cells.[25,26] Similar to CAR T-cell therapy, almost one-half of patients who receive this therapy experience cytokine release syndrome.
Mosunetuzumab
Mosunetuzumab is a bispecific T-cell engager that binds to CD20 and CD3.[25]
Evidence (mosunetuzumab):
A single-arm, multicenter, phase II study included 90 patients with relapsed or refractory follicular lymphoma who had received two or more prior lines of therapy (including an anti-CD20 therapy and an alkylating agent). Patients received mosunetuzumab.[27]
With a median follow-up of 37.4 months, the objective response rate was 77.8% (95% CI, 67.8%–85.9%), and the complete response rate was 60.0% (95% CI, 49.1%–70.2%), per investigator assessment. The median PFS (per investigator assessment) was 24.0 months. The 3-year OS rate was 82.4% (95% CI, 73.8%–91.0%).[27][Level of evidence C1]
Cytokine release syndrome occurred in 44.4% of patients; 97.2% of cases were grade 1 or 2 in severity.
Epcoritamab
Epcoritamab is a bispecific T-cell engager that binds to CD20 and CD3.
Evidence (epcoritamab):
A single-arm multicenter study (NCT03625037), included 128 patients with relapsed or refractory follicular lymphoma who had received two or more lines of therapy. Patients received epcoritamab.[28]
With a median follow-up of 17.4 months, the overall response rate was 82.0 % (95% CI, 74.3%–88.3%) and the complete response rate was 62.5% (95% CI, 53.5%–70.9%).[28][Level of evidence C3]
Only 2% of patients had grade 3 or higher cytokine release syndrome events.
Chimeric antigen receptor (CAR) T-cell therapy
CAR T-cell therapy, with the autologous anti-CD19 therapeutics axicabtagene ciloleucel (axi-cel), lisocabtagene maraleucel (liso-cel), or tisagenlecleucel (tisa-cel), has been approved for patients with relapsed follicular lymphoma after two or more lines of prior therapy.
Evidence (CAR T-cell therapy):
In a phase II trial, 159 patients with relapsed or refractory follicular lymphoma (n = 127), marginal zone lymphoma (n = 31), or diffuse large B-cell lymphoma (n = 1) received axi-cel.[29,30]
With a median follow-up of 41.7 months, the overall response rate was 92% (95% CI, 85%–97%), and the complete response rate was 74%.
The median PFS was 40.2 months for patients with follicular lymphoma and was not reached for patients with marginal zone lymphoma.[30][Level of evidence C2]
Cytokine release syndrome occurred in 78% of patients and was grade 3 or 4 in 6% of patients.
Tocilizumab was required in 50% of all patients, and 5% required vasopressors. Grade 3 or 4 neurological events occurred in 15% of patients.
In a phase II trial, 98 patients with relapsed or refractory follicular lymphoma after two or more lines of prior therapy received anti-CD19 CAR T-cell therapy with tisa-cel.[31]
With a median follow-up of 16.6 months, the complete response rate was 69.1% (95% CI, 58.8%–78.3%), and the overall response rate was 86.2% (95% CI, 77.5%–92.4%).[31][Level of evidence C3]
Grade 3 or 4 cytokine release syndrome occurred in 48.5% of patients, and 37.1% had grade 3 or 4 neurotoxicity.
A phase II trial (NCT04245839) included 130 patients with relapsed or refractory follicular lymphoma who had received two or more prior lines of therapy. Patients had high-risk features (progression of disease within 24 months after first-line chemoimmunotherapy or disease refractory to rituximab and alkylators). Patients received liso-cel.[32]
With a median follow-up of 18.9 months, the overall response rate was 97% (95% CI, 91.6%–99.4%), and the complete response rate was 94% (95% CI, 87.5%–97.8%).[32][Level of evidence C3]
Cytokine release syndrome occurred in 56% of patients and was grade 3 or higher in 1% of patients. Neurological side effects occurred in 15% of patients and was grade 3 or higher in 2% of patients.
CAR T cells are being used for high-risk patients whose disease has relapsed rapidly after chemoimmunotherapy. Such an approach is considered in the context of numerous other available agents.
Stem cell transplant
In many institutions, autologous or allogeneic SCTs are being used for high-risk patients whose disease has relapsed rapidly after chemoimmunotherapy. Such an approach is considered in the context of numerous other available agents.[33–37]
Evidence (SCT):
The German Low-Grade Lymphoma Study Group treated 307 patients with follicular lymphoma with two cycles of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone)-like induction chemotherapy and then randomly assigned them to receive autologous SCT or interferon maintenance.[38]
With a median follow-up of 4.2 years, the 5-year PFS rate was 65% for patients who received a transplant versus 33% for patients who received interferon (P < .001). There was no difference in OS.[38][Level of evidence B1]
Palliative radiation therapy
Palliation may be achieved with very low-dose (4 Gy) involved-field radiation therapy in two fractions for patients with indolent and aggressive relapsed disease.[39] In a prospective randomized trial, treatment with 4 Gy was inferior to treatment with 24 Gy in 12 fractions in PFS (77% vs. 92%, P < .0001).[40][Level of evidence B1]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
Casulo C, Byrtek M, Dawson KL, et al.: Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J Clin Oncol 33 (23): 2516-22, 2015. [PUBMED Abstract]
Weisdorf DJ, Andersen JW, Glick JH, et al.: Survival after relapse of low-grade non-Hodgkin’s lymphoma: implications for marrow transplantation. J Clin Oncol 10 (6): 942-7, 1992. [PUBMED Abstract]
Peterson BA: Current treatment of follicular low-grade lymphomas. Semin Oncol 26 (5 Suppl 14): 2-11, 1999. [PUBMED Abstract]
Haas RL, Poortmans P, de Jong D, et al.: High response rates and lasting remissions after low-dose involved field radiotherapy in indolent lymphomas. J Clin Oncol 21 (13): 2474-80, 2003. [PUBMED Abstract]
Tsimberidou AM, O’Brien S, Khouri I, et al.: Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol 24 (15): 2343-51, 2006. [PUBMED Abstract]
Montoto S, Davies AJ, Matthews J, et al.: Risk and clinical implications of transformation of follicular lymphoma to diffuse large B-cell lymphoma. J Clin Oncol 25 (17): 2426-33, 2007. [PUBMED Abstract]
Yuen AR, Kamel OW, Halpern J, et al.: Long-term survival after histologic transformation of low-grade follicular lymphoma. J Clin Oncol 13 (7): 1726-33, 1995. [PUBMED Abstract]
Link BK, Maurer MJ, Nowakowski GS, et al.: Rates and outcomes of follicular lymphoma transformation in the immunochemotherapy era: a report from the University of Iowa/MayoClinic Specialized Program of Research Excellence Molecular Epidemiology Resource. J Clin Oncol 31 (26): 3272-8, 2013. [PUBMED Abstract]
Davis TA, White CA, Grillo-López AJ, et al.: Single-agent monoclonal antibody efficacy in bulky non-Hodgkin’s lymphoma: results of a phase II trial of rituximab. J Clin Oncol 17 (6): 1851-7, 1999. [PUBMED Abstract]
Piro LD, White CA, Grillo-López AJ, et al.: Extended Rituximab (anti-CD20 monoclonal antibody) therapy for relapsed or refractory low-grade or follicular non-Hodgkin’s lymphoma. Ann Oncol 10 (6): 655-61, 1999. [PUBMED Abstract]
Davis TA, Grillo-López AJ, White CA, et al.: Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin’s lymphoma: safety and efficacy of re-treatment. J Clin Oncol 18 (17): 3135-43, 2000. [PUBMED Abstract]
Hainsworth JD, Litchy S, Shaffer DW, et al.: Maximizing therapeutic benefit of rituximab: maintenance therapy versus re-treatment at progression in patients with indolent non-Hodgkin’s lymphoma–a randomized phase II trial of the Minnie Pearl Cancer Research Network. J Clin Oncol 23 (6): 1088-95, 2005. [PUBMED Abstract]
Lockmer S, Østenstad B, Hagberg H, et al.: Chemotherapy-Free Initial Treatment of Advanced Indolent Lymphoma Has Durable Effect With Low Toxicity: Results From Two Nordic Lymphoma Group Trials With More Than 10 Years of Follow-Up. J Clin Oncol : JCO1800262, 2018. [PUBMED Abstract]
Forstpointner R, Dreyling M, Repp R, et al.: The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 104 (10): 3064-71, 2004. [PUBMED Abstract]
Canellos GP: CHOP may have been part of the beginning but certainly not the end: issues in risk-related therapy of large-cell lymphoma. J Clin Oncol 15 (5): 1713-6, 1997. [PUBMED Abstract]
van Oers MH, Van Glabbeke M, Giurgea L, et al.: Rituximab maintenance treatment of relapsed/resistant follicular non-Hodgkin’s lymphoma: long-term outcome of the EORTC 20981 phase III randomized intergroup study. J Clin Oncol 28 (17): 2853-8, 2010. [PUBMED Abstract]
van Oers MH, Klasa R, Marcus RE, et al.: Rituximab maintenance improves clinical outcome of relapsed/resistant follicular non-Hodgkin lymphoma in patients both with and without rituximab during induction: results of a prospective randomized phase 3 intergroup trial. Blood 108 (10): 3295-301, 2006. [PUBMED Abstract]
Martinelli G, Schmitz SF, Utiger U, et al.: Long-term follow-up of patients with follicular lymphoma receiving single-agent rituximab at two different schedules in trial SAKK 35/98. J Clin Oncol 28 (29): 4480-4, 2010. [PUBMED Abstract]
Sehn LH, Chua N, Mayer J, et al.: Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol 17 (8): 1081-93, 2016. [PUBMED Abstract]
Cheson BD, Chua N, Mayer J, et al.: Overall Survival Benefit in Patients With Rituximab-Refractory Indolent Non-Hodgkin Lymphoma Who Received Obinutuzumab Plus Bendamustine Induction and Obinutuzumab Maintenance in the GADOLIN Study. J Clin Oncol 36 (22): 2259-2266, 2018. [PUBMED Abstract]
Witzig TE, Wiernik PH, Moore T, et al.: Lenalidomide oral monotherapy produces durable responses in relapsed or refractory indolent non-Hodgkin’s Lymphoma. J Clin Oncol 27 (32): 5404-9, 2009. [PUBMED Abstract]
Leonard JP, Jung SH, Johnson J, et al.: Randomized Trial of Lenalidomide Alone Versus Lenalidomide Plus Rituximab in Patients With Recurrent Follicular Lymphoma: CALGB 50401 (Alliance). J Clin Oncol 33 (31): 3635-40, 2015. [PUBMED Abstract]
Zinzani PL, Mayer J, Flowers CR, et al.: ROSEWOOD: A Phase II Randomized Study of Zanubrutinib Plus Obinutuzumab Versus Obinutuzumab Monotherapy in Patients With Relapsed or Refractory Follicular Lymphoma. J Clin Oncol 41 (33): 5107-5117, 2023. [PUBMED Abstract]
Morschhauser F, Tilly H, Chaidos A, et al.: Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 21 (11): 1433-1442, 2020. [PUBMED Abstract]
Budde LE, Sehn LH, Matasar M, et al.: Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol 23 (8): 1055-1065, 2022. [PUBMED Abstract]
Hutchings M, Mous R, Clausen MR, et al.: Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet 398 (10306): 1157-1169, 2021. [PUBMED Abstract]
Sehn LH, Bartlett NL, Matasar MJ, et al.: Long-term 3-year follow-up of mosunetuzumab in relapsed or refractory follicular lymphoma after ≥2 prior therapies. Blood 145 (7): 708-719, 2025. [PUBMED Abstract]
Linton KM, Vitolo U, Jurczak W, et al.: Epcoritamab monotherapy in patients with relapsed or refractory follicular lymphoma (EPCORE NHL-1): a phase 2 cohort of a single-arm, multicentre study. Lancet Haematol 11 (8): e593-e605, 2024. [PUBMED Abstract]
Jacobson CA, Chavez JC, Sehgal AR, et al.: Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 23 (1): 91-103, 2022. [PUBMED Abstract]
Neelapu SS, Chavez JC, Sehgal AR, et al.: Three-year follow-up analysis of axicabtagene ciloleucel in relapsed/refractory indolent non-Hodgkin lymphoma (ZUMA-5). Blood 143 (6): 496-506, 2024. [PUBMED Abstract]
Fowler NH, Dickinson M, Dreyling M, et al.: Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med 28 (2): 325-332, 2022. [PUBMED Abstract]
Morschhauser F, Dahiya S, Palomba ML, et al.: Lisocabtagene maraleucel in follicular lymphoma: the phase 2 TRANSCEND FL study. Nat Med 30 (8): 2199-2207, 2024. [PUBMED Abstract]
Brice P, Simon D, Bouabdallah R, et al.: High-dose therapy with autologous stem-cell transplantation (ASCT) after first progression prolonged survival of follicular lymphoma patients included in the prospective GELF 86 protocol. Ann Oncol 11 (12): 1585-90, 2000. [PUBMED Abstract]
Khouri IF, McLaughlin P, Saliba RM, et al.: Eight-year experience with allogeneic stem cell transplantation for relapsed follicular lymphoma after nonmyeloablative conditioning with fludarabine, cyclophosphamide, and rituximab. Blood 111 (12): 5530-6, 2008. [PUBMED Abstract]
Sebban C, Brice P, Delarue R, et al.: Impact of rituximab and/or high-dose therapy with autotransplant at time of relapse in patients with follicular lymphoma: a GELA study. J Clin Oncol 26 (21): 3614-20, 2008. [PUBMED Abstract]
Thomson KJ, Morris EC, Milligan D, et al.: T-cell-depleted reduced-intensity transplantation followed by donor leukocyte infusions to promote graft-versus-lymphoma activity results in excellent long-term survival in patients with multiply relapsed follicular lymphoma. J Clin Oncol 28 (23): 3695-700, 2010. [PUBMED Abstract]
Lenz G, Dreyling M, Schiegnitz E, et al.: Myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission prolongs progression-free survival in follicular lymphoma: results of a prospective, randomized trial of the German Low-Grade Lymphoma Study Group. Blood 104 (9): 2667-74, 2004. [PUBMED Abstract]
Haas RL, Poortmans P, de Jong D, et al.: Effective palliation by low dose local radiotherapy for recurrent and/or chemotherapy refractory non-follicular lymphoma patients. Eur J Cancer 41 (12): 1724-30, 2005. [PUBMED Abstract]
Hoskin PJ, Kirkwood AA, Popova B, et al.: 4 Gy versus 24 Gy radiotherapy for patients with indolent lymphoma (FORT): a randomised phase 3 non-inferiority trial. Lancet Oncol 15 (4): 457-63, 2014. [PUBMED Abstract]
Other Lymphoproliferative and Related Disorders
Castleman Disease
A biopsy of localized or multifocal collections of lymph nodes may lead to a diagnosis of Castleman disease (CD). Strictly speaking, this uncommon diagnosis is not a lymphoma or even a malignancy. Yet, many patients with CD may be seen and treated by hematologists or oncologists.
Localized or unicentric CD is usually asymptomatic and occurs in the mediastinum, which is the most common presentation for CD.[1] Watchful waiting, surgery, or radiation therapy can be used to treat this indolent form. Multicentric CD (30% of CD patients) presents with lymphadenopathy in multiple sites; symptoms such as fever, night sweats, weight loss, and fatigue; and laboratory abnormalities such as anemia, low albumin level, elevated C-reactive protein level, and high fibrinogen level.[1] Multicentric CD (MCD) is subdivided into human herpes virus-8–associated MCD (usually with HIV or severe immunocompromise) or idiopathic MCD. Cytopenias and cytokine storm are attributed to interleukin-6 (IL-6) overproduction. MCD is a feature seen in POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin abnormalities) syndrome [2] and TAFRO (thrombocytopenia, anasarca, fever, reticulin fibrosis, and organomegaly) syndrome.[3,4] Therapy with siltuximab (an anti–IL-6 monoclonal antibody), rituximab (an anti-CD20 monoclonal antibody), or chemotherapeutic agents has been presented in anecdotal nonrandomized series.[5–8]
True Histiocytic Lymphoma
True histiocytic lymphomas are very rare tumors that show histiocytic differentiation and express histiocytic markers in the absence of B-cell or T-cell lineage-specific immunologic markers.[9,10] Care must be taken with immunophenotypic tests to exclude anaplastic large cell lymphoma or hemophagocytic syndromes caused by viral infections, especially Epstein-Barr virus.
Therapeutic options
Therapy is modeled after the treatment of comparably staged diffuse large cell lymphomas, but the optimal approach remains to be defined.
References
van Rhee F, Voorhees P, Dispenzieri A, et al.: International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 132 (20): 2115-2124, 2018. [PUBMED Abstract]
Dispenzieri A: POEMS Syndrome: 2019 Update on diagnosis, risk-stratification, and management. Am J Hematol 94 (7): 812-827, 2019. [PUBMED Abstract]
Zhang Y, Suo SS, Yang HJ, et al.: Clinical features and treatment of 7 Chinese TAFRO syndromes from 96 de novo Castleman diseases: a 10-year retrospective study. J Cancer Res Clin Oncol 146 (2): 357-365, 2020. [PUBMED Abstract]
Fujimoto S, Sakai T, Kawabata H, et al.: Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am J Hematol 94 (9): 975-983, 2019. [PUBMED Abstract]
Tonialini L, Bonfichi M, Ferrero S, et al.: Siltuximab in relapsed/refractory multicentric Castleman disease: Experience of the Italian NPP program. Hematol Oncol 36 (4): 689-692, 2018. [PUBMED Abstract]
Dong Y, Zhang L, Nong L, et al.: Effectiveness of rituximab-containing treatment regimens in idiopathic multicentric Castleman disease. Ann Hematol 97 (9): 1641-1647, 2018. [PUBMED Abstract]
Zhang L, Zhao AL, Duan MH, et al.: Phase 2 study using oral thalidomide-cyclophosphamide-prednisone for idiopathic multicentric Castleman disease. Blood 133 (16): 1720-1728, 2019. [PUBMED Abstract]
van Rhee F, Wong RS, Munshi N, et al.: Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 15 (9): 966-74, 2014. [PUBMED Abstract]
Soslow RA, Davis RE, Warnke RA, et al.: True histiocytic lymphoma following therapy for lymphoblastic neoplasms. Blood 87 (12): 5207-12, 1996. [PUBMED Abstract]
Kamel OW, Gocke CD, Kell DL, et al.: True histiocytic lymphoma: a study of 12 cases based on current definition. Leuk Lymphoma 18 (1-2): 81-6, 1995. [PUBMED Abstract]
Latest Updates to This Summary (05/14/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Revised text to state that aggressive non-Hodgkin lymphoma (NHL) has a worse prognosis in the short term, but a significant number of patients can be cured with intensive combination chemotherapy regimens. More than 70% of patients with aggressive NHL can be cured.
Revised text about the Follicular Lymphoma International Prognostic Index to state that patients with zero or one risk factor have a 10-year survival rate of 67%, and four or five risk factors confer a 10-year survival rate of 36%. Also added text about the nine prognostic variables identified by the Follicular Lymphoma Evaluation Index model.
Revised text about the results of a single-arm, multicenter, phase II study of 90 patients with relapsed or refractory follicular lymphoma who received mosunetuzumab (cited Sehn et al. as reference 27 and level of evidence C1).
Revised text about the results of a phase II trial of 159 patients with relapsed or refractory follicular lymphoma, marginal zone lymphoma, or diffuse large B-cell lymphoma who received axicabtagene ciloleucel (cited Neelapu et al. as reference 30 and level of evidence C2).
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of indolent adult B-cell non-Hodgkin lymphoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Indolent B-Cell Non-Hodgkin Lymphoma Treatment are:
Eric J. Seifter, MD (Johns Hopkins University)
Cole H. Sterling, MD (Johns Hopkins University)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Indolent B-Cell Non-Hodgkin Lymphoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/lymphoma/hp/indolent-b-cell-lymphoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 37437080]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
We offer evidence-based supportive and palliative care information for health professionals on the assessment and management of cancer-related symptoms and conditions.

. There are also two pullouts: one showing a close up of the inside structure of a lymph node and the attached artery, vein, and lymph vessels with arrows showing how the lymph (clear, watery fluid) moves into and out of the lymph node, and another showing a close up of bone marrow with blood cells.")
; left panel shows blood being drawn from a vein on the inside of the elbow using a tube attached to a syringe; right panel shows a laboratory test tube with blood cells separated into layers: plasma, white blood cells, platelets, and red blood cells.")

 being inserted into the lower back. Inset shows a close-up of the spinal needle inserted into the cerebrospinal fluid (CSF) in the lower part of the spinal column.")




 the top left panel shows cancer in the liver; (b) the top right panel shows cancer in the left lung and in two groups of lymph nodes below the diaphragm; (c) the bottom left panel shows cancer in the left lung and in a group of lymph nodes above the diaphragm and below the diaphragm; and (d) the bottom right panel shows cancer in both lungs, the liver, and the bone marrow (pullout). Also shown is primary cancer in the lymph nodes and a pullout of the brain with cerebrospinal fluid (in blue).")
 in the brain and spinal cord, and an Ommaya reservoir (a dome-shaped container that is placed under the scalp during surgery; it holds the drugs as they flow through a small tube into the brain). Top section shows a syringe and needle injecting anticancer drugs into the Ommaya reservoir. Bottom section shows a syringe and needle injecting anticancer drugs directly into the cerebrospinal fluid in the lower part of the spinal column.")