What is the PSA test?
Prostate-specific antigen, or PSA, is a protein produced by normal, as well as malignant, cells of the prostate gland. Both prostate cancer and several benign conditions (particularly benign prostatic hyperplasia, or BPH, and prostatitis) can cause PSA levels in the blood to rise.
The PSA test measures the level of PSA in the blood. This test is used in several different ways:
- to monitor the progression of prostate cancer in men who have already been diagnosed with the disease
- to follow up on prostate symptoms, such as painful or frequent urination, blood in urine or semen, and pelvic and/or back pain
- to screen for prostate cancer in men who do not have symptoms of the disease
Is the PSA test recommended for prostate cancer screening?
The PSA test is not recommended for routine prostate cancer screening in the general population. It was used for this purpose for several decades, beginning in the late 1980s. But by around 2008, as more was learned about both the benefits and harms of prostate cancer screening, many professional medical organizations began to caution against routine population screening with the PSA test. Most organizations now recommend that individuals who are considering PSA screening first discuss the risks and benefits with their doctors before making a decision.
Some organizations do recommend that men who are at higher risk of prostate cancer have routine PSA testing, beginning at age 40 or 45. Those at higher risk include Black men, men with inherited variants in BRCA2 (and to a lesser extent, in BRCA1), and men whose father or brother had prostate cancer.
The current recommendation of the United States Preventive Serves Task Force (USPSTF), which applies both to the general population and to those at increased risk due to race/ethnicity or family history, is as follows:
- For individuals aged 55 to 69 years, the decision to undergo periodic PSA-based screening for prostate cancer should be an individual one. Before making the decision, a person should discuss the potential benefits and harms of screening with their clinician and consider these in the context of their own values and preferences.
- PSA-based screening for prostate cancer is not recommended for individuals 70 years and older.
Currently, Medicare provides coverage for an annual PSA test for all Medicare-eligible individuals over 50. Many private insurers cover PSA screening as well.
What is a normal PSA test result?
There is no single threshold that distinguishes a normal versus an abnormal PSA result. This is in part because there is no specific PSA level that means that someone has prostate cancer. However, the higher someone’s PSA level, the likelier it is that prostate cancer is present.
In general, a PSA level above 4.0 ng/mL is considered abnormal and may result in a recommendation for prostate biopsy. However, because PSA levels increase with age, some doctors apply a higher cutoff (such as 5 ng/ml) for older men and a lower cutoff ( such as 2.5 ng/mL) for younger men (1).
In addition, a lower cutoff for abnormal is used in men taking certain drugs, including finasteride and dutasteride, which are used to treat BPH. These drugs lower the PSA level.
Various factors can increase someone’s PSA level temporarily. An infection or inflammation of the prostate or having had a recent prostate biopsy can cause PSA levels to be raised for a month or two. Vigorous exercise (such as cycling) and ejaculation can also increase the PSA level transiently. People are generally recommended to wait until any conditions that can change PSA level resolve before they have testing and to avoid activities that may raise the PSA level for 2 days before testing.
What is done if a screening test shows an elevated PSA level?
If someone who has no symptoms of prostate cancer chooses to undergo prostate cancer screening and is found to have an abnormal PSA level, the doctor may recommend another PSA test in 6 to 8 weeks to confirm the original finding. If the PSA level is still elevated, the doctor may recommend continued observation with repeat PSA tests along with digital rectal exams (DREs) to watch for any changes over time.
If the PSA level continues to rise—especially if it rises quickly—or if a lump is detected during a DRE, the doctor may recommend additional tests. These may include additional blood- or urine-based tests, or imaging tests, such as magnetic resonance imaging (MRI) or high-resolution micro-ultrasound.
Alternatively, the doctor may recommend a prostate biopsy without further testing. During this procedure, multiple samples of prostate tissue are collected by inserting hollow needles into the prostate and then withdrawing them. The biopsy needle may be inserted through the wall of the rectum (transrectal biopsy) or through the perineum (transperineal biopsy). A pathologist then examines the collected tissue under a microscope. Although both biopsy techniques are guided by ultrasound imaging so the doctor can view the prostate during the biopsy procedure, ultrasound cannot be used alone to diagnose prostate cancer. An MRI-guided biopsy may be performed for patients with suspicious areas seen on MRI.
What are some of the potential benefits and harms of the PSA test for prostate cancer screening?
The potential benefit of the PSA test for prostate cancer screening is that it may help detect prostate cancer earlier, before it spreads and when it may be easier to treat, possibly reducing someone’s risk of dying from prostate cancer.
A systematic review and meta-analysis of all randomized controlled trials comparing PSA screening with usual care in men without a diagnosis of prostate cancer concluded that PSA screening for prostate cancer leads to a small reduction in prostate cancer mortality over 10 years (2).
However, this potential benefit needs to be balanced against several potential harms:
- Some cancers detected through PSA screening grow so slowly that they would never cause symptoms or become life threatening. However, treating them can cause harms. Detecting tumors that would not have caused problems during someone’s lifetime is called “overdiagnosis,” and treating them is called “overtreatment.”
Overtreatment exposes a person unnecessarily to potential complications. These include urinary, bowel, and sexual side effects, such as leaking of urine following surgery; increased frequency and urgency of urination following radiation; loose stools or, less commonly, rectal bleeding, following radiation; and loss of erections or decreased erections, following both surgery and radiation.
- Detecting prostate cancer earlier does not always result in cure. While the PSA test can help detect small tumors, some of these tumors, regardless of size, may have already spread beyond the prostate before being detected and may not be curable.
- The PSA test may give false-positive results. A false-positive test result occurs when the PSA level is elevated but no cancer is present. A false-positive test result may create anxiety and lead to additional medical procedures, such as a prostate biopsy, that can be harmful. Possible side effects of biopsies include serious infections, pain, and bleeding.
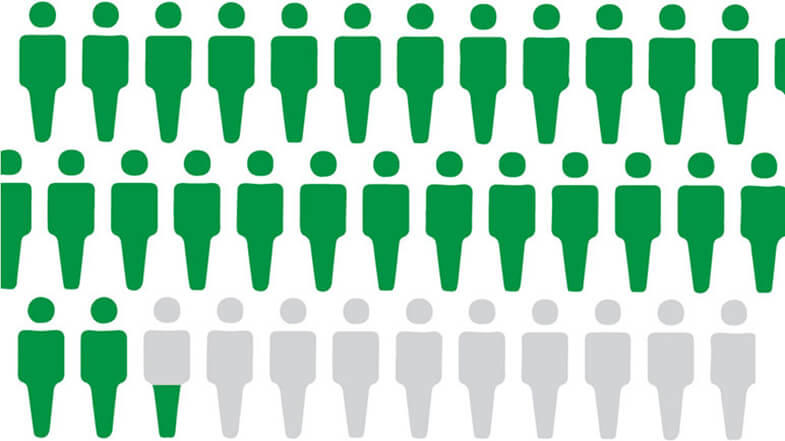
False-positive test results are common with PSA screening. About 6%–7% of men have a false-positive PSA test on any given screening round, and only about 25% of men who have a biopsy due to an elevated PSA level are found to have prostate cancer (3).
The United States Preventive Services Task Force has estimated that, for every 1,000 men ages 55 to 69 years who are screened for 13 years (4):
- About 1.3 deaths from prostate cancer would be avoided (or 1 death avoided per 769 men screened). Subsequent trial data showed that up to 2 deaths from prostate cancer would be avoided per every 1,000 men screened (or 1 death avoided in 570 men screened) (5).
- 3 men would avoid developing metastatic cancer.
- 5 men would die from prostate cancer despite having screening, diagnosis, and treatment.
- 240 men would have a positive PSA test result, many of whom would have a biopsy that shows that the result was a false-positive; some men who had a biopsy would experience at least moderately bothersome symptoms (pain, bleeding, or infection) from the procedure (and 2 would be hospitalized).
- 100 men would be diagnosed with prostate cancer. Of those, 80 would be treated (either immediately or after a period of active surveillance) with surgery or radiation. Many of these men would have a serious complication from treatment, with 50 experiencing sexual dysfunction and 15 experiencing urinary incontinence.
- 200 men would die of causes other than prostate cancer.
How is the PSA test used in people who have been treated for prostate cancer?
The PSA test is used to monitor people after surgery or radiation therapy for prostate cancer to see if their cancer has recurred (come back). If a person’s PSA level begins to rise after prostate cancer treatment, it may be the first sign of a recurrence. Such a “biochemical relapse” typically appears months or years before the recurrence causes symptoms.
However, a single elevated PSA measurement in someone who has a history of prostate cancer does not always mean that the cancer has come back. Someone who has been treated for prostate cancer should discuss an elevated PSA level with their doctor. The doctor may recommend repeating the PSA test or performing other tests to check for evidence of a recurrence. The doctor may look for a trend of rising PSA level over time rather than a single elevated PSA level.
A rising trend in PSA level over time in combination with other findings, such as an abnormal result on imaging tests, may lead the doctor to recommend further cancer treatment.
How are researchers trying to improve the PSA test?
Scientists are investigating ways to improve the PSA test and to identify other potential biomarkers and imaging tests to help doctors better distinguish cancerous from benign conditions and slow-growing cancers from fast-growing, potentially lethal cancers. None of these tests has yet been proven to decrease the risk of death from prostate cancer. Some of the methods being studied include:
Blood-based tests. Tests that measure different characteristics of PSA in the blood may help
- determine whether a prostate biopsy is needed (Prostate Health Index)
- determine the risk of a high-grade prostate cancer requiring a biopsy (IsoPSA [6])
- assess the risk of aggressive prostate cancer in someone with an abnormal prostate screening result (4Kscore test)
Urine-based tests. Tests that measure biomarkers in the urine may help
- prevent an unnecessary biopsy among people with an elevated blood PSA (PCA3 mRNA and the TMPRSS2-ERG gene fusion in combination with PSA and the MPS2 test [7])
- screen for prostate cancer (exosomal PCA3, SPDEF, and ERG RNA [ExoDx Prostate IntelliScore]; HOXC6 and DLX1 mRNA after an abnormal PSA and/or DRE [SelectMDx]; and small non-coding RNAs [Sentinel PCa Test])
Imaging tests. Tests that integrate magnetic resonance imaging (MRI) into PSA and biomarker screening are being studied to assess the risk of prostate cancer before a biopsy (8).

. Panel on the left shows the normal prostate and flow of urine from the bladder through the urethra. Panel on the right shows an enlarged prostate pressing on the bladder and urethra, blocking the flow of urine.")