
Primary Liver Cancer Treatment (PDQ®)–Health Professional Version
General Information About Primary Liver Cancer
Liver cancer includes two major types: hepatocellular carcinoma (HCC) and intrahepatic bile duct cancer. For information about bile duct cancer, see Bile Duct Cancer (Cholangiocarcinoma) Treatment. For more information about other, less common types of liver cancer, see the Cellular Classification of Primary Liver Cancer section.
Incidence and Mortality
Estimated new cases and deaths from liver and intrahepatic bile duct cancer in the United States in 2025:[1]
- New cases: 42,240.
- Deaths: 30,090.
HCC is relatively uncommon in the United States, although its incidence is rising, principally in relation to the spread of hepatitis C virus infection.[2] Worldwide, HCC is the sixth most prevalent cancer and the third leading cause of cancer-related deaths.[3]
Anatomy

Risk Factors
Increasing age is the most important risk factor for most cancers. Other risk factors for liver (hepatocellular) cancer include:
For more information, see Liver (Hepatocellular) Cancer Prevention.
Screening
For more information, see Liver (Hepatocellular) Cancer Screening.
Diagnostic Factors
Lesions smaller than 1 cm that are detected during screening in patients at high risk of HCC do not require further diagnostic evaluation. Most of these lesions will be cirrhotic lesions rather than HCC.[24][Level of evidence C1] Close follow-up at 3-month intervals is a common surveillance strategy, using the same technique that first documented the presence of the lesions.
For patients with liver lesions larger than 1 cm who are at risk of HCC, a diagnosis can be considered. The tests required to diagnose HCC may include imaging, biopsy, or both.
Diagnostic imaging
In patients with cirrhosis, liver disease, or other risk factors for HCC, and with lesions greater than 1 cm, triple-phase, contrast-enhanced studies (dynamic computed tomography [CT] or magnetic resonance imaging [MRI]) can be used to diagnose HCC.[25]
A triple-phase CT or MRI assesses the entire liver in distinct phases of perfusion. Following the controlled administration of intravenous contrast media, the arterial and venous phases of perfusion are imaged.
During the arterial phase of the study, HCC enhances more intensely than the surrounding liver because the arterial blood in the liver is diluted by venous blood that does not contain contrast, whereas the HCC contains only arterial blood. In the venous phase, the HCC enhances less than the surrounding liver (which is referred to as the venous washout of HCC), because the arterial blood flowing through the lesion no longer contains contrast; however, the portal blood in the liver now contains contrast.
The presence of arterial uptake followed by washout in a single dynamic study is highly specific (95%–100%) for HCC of 1 to 3 cm in diameter and virtually diagnostic of HCC.[26–28][Level of evidence C1] In these cases, the diagnosis of HCC may be established without a second imaging modality, even in the absence of a biopsy confirmation.[28–30][Level of evidence C1]
However, if a first imaging modality, such as a contrast-enhanced CT or MRI, is not conclusive, sequential imaging with a different modality can improve sensitivity for HCC detection (from 33% to 41% for either CT or MRI to 76% for both studies when performed sequentially) without a decrease in specificity.[27]
If, despite the use of two imaging modalities, a lesion larger than 1 cm remains uncharacterized in a patient at high risk of HCC (i.e., with no or only one classic enhancement pattern), a liver biopsy can be considered.[28,29]
Liver biopsy
A liver biopsy may be performed when a diagnosis of HCC is not established by a dynamic imaging modality (three-phase CT or MRI) for liver lesions 1 cm or larger in high-risk patients.
Alpha-fetoprotein (AFP) levels
AFP is insufficiently sensitive or specific for use as a diagnostic assay. AFP can be elevated in intrahepatic cholangiocarcinoma and in some cases in which there are metastases from colon cancer. Finding a mass in the liver of a patient with an elevated AFP does not automatically indicate HCC. However, if the AFP level is high, it can be used to monitor for recurrence.
Prognosis
The natural course of early tumors is poorly understood because most HCC patients receive treatment. However, older reports have described 3-year survival rates of 13% to 21% in patients who do not receive any specific treatment.[31,32] At present, only 10% to 23% of patients with HCC may be surgical candidates for curative-intent treatment.[33,34] The 5-year overall survival (OS) rate for patients with early HCC who undergo liver transplant is 44% to 78%. For patients who undergo a liver resection, the OS rate is 27% to 70%.[35]
Liver transplant, surgical resection, and ablation offer high rates of complete responses and a potential for cure in patients with early HCC.[29]
The natural course of advanced-stage HCC is better known. Untreated patients with advanced disease usually survive less than 6 months.[36] The survival rate of untreated patients in 25 randomized clinical trials ranged from 10% to 72% at 1 year and 8% to 50% at 2 years.[37]
Unlike most patients with solid tumors, the prognosis of patients with HCC is affected by the tumor stage at presentation and by the underlying liver function. The following prognostic factors guide the selection of treatment:
References
- American Cancer Society: Cancer Facts and Figures 2025. American Cancer Society, 2025. Available online. Last accessed January 16, 2025.
- Altekruse SF, McGlynn KA, Reichman ME: Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 27 (9): 1485-91, 2009. [PUBMED Abstract]
- Forner A, Llovet JM, Bruix J: Hepatocellular carcinoma. Lancet 379 (9822): 1245-55, 2012. [PUBMED Abstract]
- Bosetti C, Turati F, La Vecchia C: Hepatocellular carcinoma epidemiology. Best Pract Res Clin Gastroenterol 28 (5): 753-70, 2014. [PUBMED Abstract]
- El-Serag HB: Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 142 (6): 1264-1273.e1, 2012. [PUBMED Abstract]
- El-Serag HB, Kanwal F: Epidemiology of hepatocellular carcinoma in the United States: where are we? Where do we go? Hepatology 60 (5): 1767-75, 2014. [PUBMED Abstract]
- Lafaro KJ, Demirjian AN, Pawlik TM: Epidemiology of hepatocellular carcinoma. Surg Oncol Clin N Am 24 (1): 1-17, 2015. [PUBMED Abstract]
- Fattovich G, Stroffolini T, Zagni I, et al.: Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 127 (5 Suppl 1): S35-50, 2004. [PUBMED Abstract]
- Grewal P, Viswanathen VA: Liver cancer and alcohol. Clin Liver Dis 16 (4): 839-50, 2012. [PUBMED Abstract]
- London WT, McGlynn K: Liver cancer. In: Schottenfeld D, Fraumeni JF Jr, eds.: Cancer Epidemiology and Prevention. 3rd ed. Oxford University Press, 2006, pp 763-86.
- McGlynn KA, Petrick JL, London WT: Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis 19 (2): 223-38, 2015. [PUBMED Abstract]
- Liu Y, Wu F: Global burden of aflatoxin-induced hepatocellular carcinoma: a risk assessment. Environ Health Perspect 118 (6): 818-24, 2010. [PUBMED Abstract]
- Chen JG, Egner PA, Ng D, et al.: Reduced aflatoxin exposure presages decline in liver cancer mortality in an endemic region of China. Cancer Prev Res (Phila) 6 (10): 1038-45, 2013. [PUBMED Abstract]
- Baffy G, Brunt EM, Caldwell SH: Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol 56 (6): 1384-91, 2012. [PUBMED Abstract]
- Diehl AM, Day C: Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N Engl J Med 377 (21): 2063-2072, 2017. [PUBMED Abstract]
- White DL, Kanwal F, El-Serag HB: Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol 10 (12): 1342-1359.e2, 2012. [PUBMED Abstract]
- Ascha MS, Hanouneh IA, Lopez R, et al.: The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 51 (6): 1972-8, 2010. [PUBMED Abstract]
- Chuang SC, Lee YC, Hashibe M, et al.: Interaction between cigarette smoking and hepatitis B and C virus infection on the risk of liver cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev 19 (5): 1261-8, 2010. [PUBMED Abstract]
- Lee YC, Cohet C, Yang YC, et al.: Meta-analysis of epidemiologic studies on cigarette smoking and liver cancer. Int J Epidemiol 38 (6): 1497-511, 2009. [PUBMED Abstract]
- Koh WP, Robien K, Wang R, et al.: Smoking as an independent risk factor for hepatocellular carcinoma: the Singapore Chinese Health Study. Br J Cancer 105 (9): 1430-5, 2011. [PUBMED Abstract]
- Lomas DA, Evans DL, Finch JT, et al.: The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 357 (6379): 605-7, 1992. [PUBMED Abstract]
- Huster D: Wilson disease. Best Pract Res Clin Gastroenterol 24 (5): 531-9, 2010. [PUBMED Abstract]
- Pfeiffenberger J, Mogler C, Gotthardt DN, et al.: Hepatobiliary malignancies in Wilson disease. Liver Int 35 (5): 1615-22, 2015. [PUBMED Abstract]
- Furuya K, Nakamura M, Yamamoto Y, et al.: Macroregenerative nodule of the liver. A clinicopathologic study of 345 autopsy cases of chronic liver disease. Cancer 61 (1): 99-105, 1988. [PUBMED Abstract]
- Brunello F, Cantamessa A, Gaia S, et al.: Radiofrequency ablation: technical and clinical long-term outcomes for single hepatocellular carcinoma up to 30 mm. Eur J Gastroenterol Hepatol 25 (7): 842-9, 2013. [PUBMED Abstract]
- Leoni S, Piscaglia F, Golfieri R, et al.: The impact of vascular and nonvascular findings on the noninvasive diagnosis of small hepatocellular carcinoma based on the EASL and AASLD criteria. Am J Gastroenterol 105 (3): 599-609, 2010. [PUBMED Abstract]
- Khalili K, Kim TK, Jang HJ, et al.: Optimization of imaging diagnosis of 1-2 cm hepatocellular carcinoma: an analysis of diagnostic performance and resource utilization. J Hepatol 54 (4): 723-8, 2011. [PUBMED Abstract]
- Sangiovanni A, Manini MA, Iavarone M, et al.: The diagnostic and economic impact of contrast imaging techniques in the diagnosis of small hepatocellular carcinoma in cirrhosis. Gut 59 (5): 638-44, 2010. [PUBMED Abstract]
- Bruix J, Sherman M; American Association for the Study of Liver Diseases: Management of hepatocellular carcinoma: an update. Hepatology 53 (3): 1020-2, 2011. [PUBMED Abstract]
- Khalili K, Kim TK, Jang HJ, et al.: Implementation of AASLD hepatocellular carcinoma practice guidelines in North America: two years of experience. [Abstract] Hepatology 48 (Suppl 1): A-128, 362A, 2008.
- Barbara L, Benzi G, Gaiani S, et al.: Natural history of small untreated hepatocellular carcinoma in cirrhosis: a multivariate analysis of prognostic factors of tumor growth rate and patient survival. Hepatology 16 (1): 132-7, 1992. [PUBMED Abstract]
- Ebara M, Ohto M, Shinagawa T, et al.: Natural history of minute hepatocellular carcinoma smaller than three centimeters complicating cirrhosis. A study in 22 patients. Gastroenterology 90 (2): 289-98, 1986. [PUBMED Abstract]
- Shah SA, Smith JK, Li Y, et al.: Underutilization of therapy for hepatocellular carcinoma in the medicare population. Cancer 117 (5): 1019-26, 2011. [PUBMED Abstract]
- Sonnenday CJ, Dimick JB, Schulick RD, et al.: Racial and geographic disparities in the utilization of surgical therapy for hepatocellular carcinoma. J Gastrointest Surg 11 (12): 1636-46; discussion 1646, 2007. [PUBMED Abstract]
- Dhir M, Lyden ER, Smith LM, et al.: Comparison of outcomes of transplantation and resection in patients with early hepatocellular carcinoma: a meta-analysis. HPB (Oxford) 14 (9): 635-45, 2012. [PUBMED Abstract]
- Okuda K, Ohtsuki T, Obata H, et al.: Natural history of hepatocellular carcinoma and prognosis in relation to treatment. Study of 850 patients. Cancer 56 (4): 918-28, 1985. [PUBMED Abstract]
- Llovet JM, Bruix J: Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology 37 (2): 429-42, 2003. [PUBMED Abstract]
- Llovet JM, Brú C, Bruix J: Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis 19 (3): 329-38, 1999. [PUBMED Abstract]
- A new prognostic system for hepatocellular carcinoma: a retrospective study of 435 patients: the Cancer of the Liver Italian Program (CLIP) investigators. Hepatology 28 (3): 751-5, 1998. [PUBMED Abstract]
Cellular Classification of Primary Liver Cancer
Malignant primary tumors of the liver consist of two major cell types, hepatocellular (90% of cases) and cholangiocarcinoma.[1]
Histological classification is as follows:
- Hepatocellular carcinoma (HCC; liver cell carcinoma).
- Fibrolamellar variant of HCC.
It is important to distinguish between the fibrolamellar variant of HCC and HCC itself because an increased proportion of patients with the fibrolamellar variant may be cured if the tumor can be resected. Found more frequently in young women, this variant generally exhibits a slower clinical course than the more common HCC.[2]
- Cholangiocarcinoma (intrahepatic bile duct carcinoma).
- Mixed hepatocellular cholangiocarcinoma.
- Undifferentiated.
- Hepatoblastoma. This occurs more often in children than in adults. For more information, see Childhood Liver Cancer Treatment.
References
- Llovet JM, Burroughs A, Bruix J: Hepatocellular carcinoma. Lancet 362 (9399): 1907-17, 2003. [PUBMED Abstract]
- Mavros MN, Mayo SC, Hyder O, et al.: A systematic review: treatment and prognosis of patients with fibrolamellar hepatocellular carcinoma. J Am Coll Surg 215 (6): 820-30, 2012. [PUBMED Abstract]
Stage Information for Primary Liver Cancer
Prognostic modeling in hepatocellular carcinoma (HCC) is complex because cirrhosis is involved in as many as 80% of cases. Tumor features and the factors related to functional hepatic reserve must be considered. The key prognostic factors are only partially known and vary at different stages of the disease.
More than ten classifications are used throughout the world, but no system is accepted worldwide. New classifications have been proposed to overcome the difficulties of having several staging systems.
This summary discusses the following three staging systems:
Barcelona Clinic Liver Cancer (BCLC) Staging System
Currently, the BCLC staging classification is the most accepted staging system for HCC and is useful in the staging of early tumors. Evidence from an American cohort has shown that BCLC staging offers better prognostic stratification power than other staging systems.[1]
The BCLC staging system attempts to overcome the limitations of previous staging systems by including variables related to the following:[2]
- Tumor stage.
- Functional status of the liver.
- Physical status.
- Cancer-related symptoms.
Five stages (0 and A through D) are identified based on the variables mentioned above. The BCLC staging system links each HCC stage to appropriate treatment modalities as follows:
- Patients with early-stage HCC may benefit from curative therapies (i.e., liver transplant, surgical resection, and radiofrequency ablation).
- Patients with intermediate-stage or advanced-stage disease may benefit from palliative treatments (i.e., transcatheter arterial chemoembolization and sorafenib).
- Patients with end-stage disease who have a very poor life expectancy are offered supportive care and palliation.
Okuda Staging System
The Okuda staging system has been extensively used in the past and includes variables related to tumor burden and liver function, such as bilirubin, albumin, and ascites. However, many significant prognostic tumor factors confirmed in both surgical and nonsurgical series (e.g., unifocal or multifocal, vascular invasion, portal venous thrombosis, or locoregional lymph node involvement) are not included.[3,4] As a result, Okuda staging is unable to stratify prognosis for early-stage cancers and mostly serves to recognize end-stage disease.
AJCC Staging System and TNM Definitions
The TNM (tumor, node, metastasis) classification for staging, proposed by the AJCC, is not widely used for liver cancer. Clinical use of TNM staging is limited because liver function is not considered. It is also difficult to use this system to select treatment options because TNM staging relies on detailed histopathological examination available only after tumor excision. TNM may be useful in prognostic prediction after liver resection.[5]
| Stage | TNM | Description |
|---|---|---|
| Tumor = primary tumor; N = regional lymph nodes; M = distant metastasis. | ||
| aReprinted with permission from AJCC: Liver. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 287–93. | ||
| IA | T1a, N0, M0 | T1a = Solitary tumor ≤2 cm. |
| N0 = No regional lymph node metastasis. | ||
| M0 = No distant metastasis. | ||
| IB | T1b, N0, M0 | T1b = Solitary tumor >2 cm without vascular invasion. |
| N0 = No regional lymph node metastasis. | ||
| M0 = No distant metastasis. | ||
| Stage | TNM | Description |
|---|---|---|
| T = primary tumor; N = regional lymph nodes; M = distant metastasis. | ||
| aReprinted with permission from AJCC: Liver. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 287–93. | ||
| II | T2, N0, M0 | T2 = Solitary tumor >2 cm with vascular invasion, or multiple tumors, none >5 cm. |
| N0 = No regional lymph node metastasis. | ||
| M0 = No distant metastasis. | ||
| Stage | TNM | Description |
|---|---|---|
| T = primary tumor; N = regional lymph nodes; M = distant metastasis. | ||
| aReprinted with permission from AJCC: Liver. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 287–93. | ||
| IIIA | T3, N0, M0 | T3 = Multiple tumors, at least one of which is >5 cm. |
| N0 = No regional lymph node metastasis. | ||
| M0 = No distant metastasis. | ||
| IIIB | T4, N0, M0 | T4 = Single tumor or multiple tumors of any size involving a major branch of the portal vein or hepatic vein, or tumor(s) with direct invasion of adjacent organs other than the gallbladder or with perforation of visceral peritoneum. |
| N0 = No regional lymph node metastasis. | ||
| M0 = No distant metastasis. | ||
| Stage | TNM | Description |
|---|---|---|
| T = primary tumor; N = regional lymph nodes; M = distant metastasis. | ||
| aReprinted with permission from AJCC: Liver. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 287–93. | ||
| IVA | Any T, N1, M0 | TX = Primary tumor cannot be assessed. |
| T0 = No evidence of primary tumor. | ||
| T1 = Solitary tumor ≤2 cm, or >2 cm without vascular invasion. | ||
| –T1a = Solitary tumor ≤2 cm. | ||
| –T1b = Solitary tumor >2 cm without vascular invasion. | ||
| T2 = Solitary tumor >2 cm with vascular invasion, or multiple tumors, none >5 cm. | ||
| T3 = Multiple tumors, at least one of which is >5 cm. | ||
| T4 = Single tumor or multiple tumors of any size involving a major branch of the portal vein or hepatic vein, or tumor(s) with direct invasion of adjacent organs other than the gallbladder or with perforation of visceral peritoneum. | ||
| N1 = Regional lymph node metastasis. | ||
| M0 = No distant metastasis. | ||
| IVB | Any T, Any N, M1 | Any T = See descriptions above in this table, stage IVA, Any T, N1, M0. |
| NX = Regional lymph nodes cannot be assessed. | ||
| N0 = No regional lymph node metastasis. | ||
| N1 = Regional lymph node metastasis. | ||
| M1 = Distant metastasis. | ||
References
- Marrero JA, Fontana RJ, Barrat A, et al.: Prognosis of hepatocellular carcinoma: comparison of 7 staging systems in an American cohort. Hepatology 41 (4): 707-16, 2005. [PUBMED Abstract]
- Llovet JM, Brú C, Bruix J: Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis 19 (3): 329-38, 1999. [PUBMED Abstract]
- Poon RT, Ng IO, Fan ST, et al.: Clinicopathologic features of long-term survivors and disease-free survivors after resection of hepatocellular carcinoma: a study of a prospective cohort. J Clin Oncol 19 (12): 3037-44, 2001. [PUBMED Abstract]
- Pompili M, Rapaccini GL, Covino M, et al.: Prognostic factors for survival in patients with compensated cirrhosis and small hepatocellular carcinoma after percutaneous ethanol injection therapy. Cancer 92 (1): 126-35, 2001. [PUBMED Abstract]
- Liver. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 287–93.
Treatment Option Overview for Primary Liver Cancer
There is no agreement on a single treatment strategy for patients with hepatocellular carcinoma (HCC). Selection of treatment is complex due to several factors, including:
- Underlying liver function.
- Extent and location of the tumor.
- General condition of the patient.
Several treatments for HCC are associated with long-term survival, including surgical resection, liver transplant, and ablation. There are no large, robust, randomized studies that compare treatments considered effective for early-stage disease, nor are there studies comparing these treatments with best supportive care. Often, patients with HCC are evaluated by a multidisciplinary team that includes hepatologists, radiologists, interventional radiologists, radiation oncologists, transplant surgeons, surgical oncologists, pathologists, and medical oncologists.
Best survivals are achieved when the HCC can be removed either by surgical resection or liver transplant. Surgical resection is usually performed in patients with localized HCC and enough functional hepatic reserve.
For patients with decompensated cirrhosis and a solitary lesion (<5 cm) or early multifocal disease (≤3 lesions, ≤3 cm in diameter), the best option is liver transplant.[1] However, the limited availability of liver donors restricts the use of this approach.
Transarterial chemoembolization, multikinase inhibitors, and immunotherapy are noncurative treatments for HCC that improve survival.[2–4]
Table 5 shows the standard treatment options for HCC.
| Stage | Treatment Options |
|---|---|
| Localized | Surveillance |
| Surgical resection | |
| Liver transplant | |
| Ablation | |
| Radiation therapy | |
| Locally advanced or metastatic | Transarterial embolization and transcatheter arterial chemoembolization in patients with nonmetastatic disease |
| First-line systemic therapy | |
| Second-line systemic therapy | |
| Radiation therapy | |
| Recurrent (liver-limited disease without vascular involvement) | Liver transplant |
| Surgical resection | |
| Ablation | |
| Radiation therapy | |
| Recurrent (extrahepatic disease or vascular involvement) | Palliative therapy |
References
- Bruix J, Sherman M; American Association for the Study of Liver Diseases: Management of hepatocellular carcinoma: an update. Hepatology 53 (3): 1020-2, 2011. [PUBMED Abstract]
- Llovet JM, Ricci S, Mazzaferro V, et al.: Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359 (4): 378-90, 2008. [PUBMED Abstract]
- Llovet JM, Bruix J: Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology 37 (2): 429-42, 2003. [PUBMED Abstract]
- Cammà C, Schepis F, Orlando A, et al.: Transarterial chemoembolization for unresectable hepatocellular carcinoma: meta-analysis of randomized controlled trials. Radiology 224 (1): 47-54, 2002. [PUBMED Abstract]
Treatment of Localized Primary Liver Cancer
About 30% of hepatocellular carcinoma (HCC) cases present as localized disease, with a solitary mass in part of the liver or as a limited number of tumors (≤3 lesions, ≤3 cm in diameter) without major vascular invasion.
Treatment Options for Localized Primary Liver Cancer
Treatment options for localized primary liver cancer include:
Resection and transplant achieve the best outcomes in well-selected candidates, and are usually considered the first option for curative intent.
Surveillance
Surveillance is an option for patients at high risk of HCC with lesions smaller than 1 cm detected during screening.[1][Level of evidence C1] Close follow-up at 3-month intervals is a common surveillance strategy, using the same technique that first documented the presence of the lesions.
Surgical resection
Surgery is the mainstay of HCC treatment.
Preoperative assessment includes three-phase helical computed tomography, magnetic resonance imaging, or both to determine the presence of an extension of a tumor across interlobar planes and potential involvement of the hepatic hilus, hepatic veins, and inferior vena cava. Tumors can be resected only if enough liver parenchyma can be spared with adequate vascular and biliary inflow and outflow. Patients with well-compensated cirrhosis can generally tolerate resection of up to 50% of their liver parenchyma.
Surgical resection can be considered for patients who meet the following criteria:
- A solitary mass.
- Good performance status.
- Normal or minimally abnormal liver function tests.
- No evidence of portal hypertension.
- No evidence of cirrhosis beyond Child-Pugh class A.
After considering the location and number of tumors and the patient’s hepatic function, only 5% to 10% of patients with liver cancer will prove to have localized disease amenable to resection.[2–6]
The principles of surgical resection involve obtaining a clear margin around the tumor, which may require any of the following procedures:
- Segmental resection.
- Hormone-lymphatic lobectomy.
- Extended lobectomy.
The 5-year overall survival (OS) rate after curative resection ranges between 27% and 70% and depends on tumor stage and underlying liver function.[2–6]
In patients with limited multifocal disease, hepatic resection is controversial.
Liver transplant
Liver transplant is a potentially curative therapy for HCC and has the benefit of treating the underlying cirrhosis, but the scarcity of organ donors limits the availability of this treatment modality.[2]
According to the Milan criteria, patients with a single HCC lesion smaller than 5 cm, or 2 to 3 lesions smaller than 3 cm are eligible for liver transplant. Expansion of the accepted transplant criteria for HCC is not supported by consistent data. Liver transplant is considered if resection is precluded because of multiple small tumor lesions (≤3 lesions, each ≤3 cm), or impaired liver function (Child-Pugh class B and class C). In patients who meet the criteria, transplant is associated with a 5-year OS rate of approximately 70%.[7][Level of evidence C1]
Ablation
When tumor excision, either by transplant or resection, is not feasible or advisable, ablation may be used if the tumor can be accessed percutaneously or, if necessary, through minimally invasive or open surgery. Ablation may be particularly useful for patients with early-stage HCC that is centrally located in the liver and cannot be surgically removed without excessive sacrifice of functional parenchyma.
Ablation can be achieved in the following ways:
- Change in temperature (e.g., radiofrequency ablation [RFA], microwave, or cryoablation).
- Exposure to a chemical substance (e.g., percutaneous ethanol injection [PEI]).
- Direct damage of the cellular membrane (definitive electroporation).
With ablation, a margin of normal liver around the tumor can be considered. Ablation is relatively contraindicated for lesions near bile ducts, the diaphragm, or other intra-abdominal organs that might be injured during the procedure. Furthermore, when tumors are located adjacent to major vessels, the blood flow in the vessels may keep thermal ablation techniques, such as RFA, from reaching optimal temperatures. This is known as the heat-sink effect, which may preclude complete tumor necrosis.
RFA achieves best results in patients with tumors smaller than 3 cm. In this subpopulation, 5-year OS rates may be as high as 59%, and the recurrence-free survival rates may not differ significantly from treatment with hepatic resection.[8,9] Local control success progressively diminishes as the tumor size increases beyond 3 cm.
PEI yields good results in patients with Child-Pugh class A cirrhosis and a single tumor smaller than 3 cm in diameter. In those cases, the 5-year OS rate can be as high as 40% to 59%.[10,11][Level of evidence C2]
In the few randomized controlled trials that included patients with Child-Pugh class A cirrhosis, RFA proved superior to PEI in rates of complete response and local recurrences. Some of those studies have also shown improved OS with RFA. Furthermore, RFA requires fewer treatment sessions than PEI to achieve comparable outcomes.[12–15]
Of note, RFA may have higher complication rates than PEI,[13] but both techniques are associated with lower complication rates than excision procedures. RFA is a well-established technique in the treatment of HCC.
Radiation therapy
Radiation therapy can be delivered with curative or palliative intent for patients with primary liver cancer. One form of radiation, stereotactic body radiation therapy (SBRT), treats patients with a small number of fractions of precise, image-guided radiation therapy at a high biologically equivalent dose. Numerous retrospective studies have shown excellent local control for patients with HCC who receive SBRT (local control rates ranging from 70%–95% at 2 years for smaller HCCs).
Evidence (curative radiation therapy):
- The phase III NRG/RTOG 1112 study (NCT01730937) evaluated sorafenib alone or SBRT followed by sorafenib in patients with HCC. Patients were included if they had Child-Pugh class A, Barcelona Clinic Liver Cancer stage B or C, new or recurrent HCC. Patients also had five or fewer lesions, a tumor sum measuring 20 cm or less, and distant metastases measuring 3 cm or less. A total of 177 patients were randomly assigned (92 to sorafenib alone, 85 to SBRT followed by sorafenib). The primary end point was OS.[16][Level of evidence B1]
- The median OS was 12.3 months for the sorafenib-alone group and 15.8 months for the SBRT-plus-sorafenib group (hazard ratio [HR], 0.77; one-sided P = .0554). This was not statistically significant.
- Grade 3 or higher adverse events were not significantly different between the two groups: 42% for the sorafenib-alone group and 47% for the SBRT-plus-sorafenib group (P = .52).
- There were three grade 5 adverse events: one hepatic failure and one death not otherwise specified in the sorafenib-alone group and one grade 5 lung infection in the SBRT-plus-sorafenib group.
Based on these results, SBRT is a standard of care treatment with curative intent for HCC. It can also be used to provide local control before liver transplant.
Evidence (radiation therapy for palliation):
- The Canadian Cancer Trials Group HE.1 trial (NCT02511522), a phase III study published in abstract form, included 66 patients with HCC. Patients were randomly assigned to receive either best supportive care alone or palliative radiation therapy to the liver (8 Gy in one fraction). Patients had end-stage disease unsuitable for local, regional, or systemic therapies. Patients also had to be more than 4 weeks from receiving chemotherapy or transcatheter arterial chemoembolization, more than 2 weeks from receiving targeted therapy or immunotherapy, and not planning any systemic therapy. The primary outcome of the study was the proportion of patients who reported an improvement of at least two points from baseline on the Brief Pain Inventory when asked to rate their liver cancer pain “intensity at worst.” A secondary end point included 3-month OS.[17][Level of evidence B3]
- A significant improvement in the “worst” pain score from baseline to 1 month was seen in 67% of patients who received radiation therapy and 22% of patients who received best supportive care.
- Although radiation therapy has not been historically used in this patient population, this study showed no decrease in OS for patients who received radiation. The 3-month OS rate was 51% for patients who received radiation therapy and 33% for patients who received best supportive care alone (P = .07), despite the study including patients with Child-Pugh class A, B, and C cirrhosis.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Furuya K, Nakamura M, Yamamoto Y, et al.: Macroregenerative nodule of the liver. A clinicopathologic study of 345 autopsy cases of chronic liver disease. Cancer 61 (1): 99-105, 1988. [PUBMED Abstract]
- Llovet JM, Fuster J, Bruix J: Intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: resection versus transplantation. Hepatology 30 (6): 1434-40, 1999. [PUBMED Abstract]
- Chok KS, Ng KK, Poon RT, et al.: Impact of postoperative complications on long-term outcome of curative resection for hepatocellular carcinoma. Br J Surg 96 (1): 81-7, 2009. [PUBMED Abstract]
- Kianmanesh R, Regimbeau JM, Belghiti J: Selective approach to major hepatic resection for hepatocellular carcinoma in chronic liver disease. Surg Oncol Clin N Am 12 (1): 51-63, 2003. [PUBMED Abstract]
- Poon RT, Fan ST, Lo CM, et al.: Long-term survival and pattern of recurrence after resection of small hepatocellular carcinoma in patients with preserved liver function: implications for a strategy of salvage transplantation. Ann Surg 235 (3): 373-82, 2002. [PUBMED Abstract]
- Dhir M, Lyden ER, Smith LM, et al.: Comparison of outcomes of transplantation and resection in patients with early hepatocellular carcinoma: a meta-analysis. HPB (Oxford) 14 (9): 635-45, 2012. [PUBMED Abstract]
- Hemming AW, Cattral MS, Reed AI, et al.: Liver transplantation for hepatocellular carcinoma. Ann Surg 233 (5): 652-9, 2001. [PUBMED Abstract]
- Huang J, Hernandez-Alejandro R, Croome KP, et al.: Radiofrequency ablation versus surgical resection for hepatocellular carcinoma in Childs A cirrhotics-a retrospective study of 1,061 cases. J Gastrointest Surg 15 (2): 311-20, 2011. [PUBMED Abstract]
- Zhou YM, Shao WY, Zhao YF, et al.: Meta-analysis of laparoscopic versus open resection for hepatocellular carcinoma. Dig Dis Sci 56 (7): 1937-43, 2011. [PUBMED Abstract]
- Huang GT, Lee PH, Tsang YM, et al.: Percutaneous ethanol injection versus surgical resection for the treatment of small hepatocellular carcinoma: a prospective study. Ann Surg 242 (1): 36-42, 2005. [PUBMED Abstract]
- Yamamoto J, Okada S, Shimada K, et al.: Treatment strategy for small hepatocellular carcinoma: comparison of long-term results after percutaneous ethanol injection therapy and surgical resection. Hepatology 34 (4 Pt 1): 707-13, 2001. [PUBMED Abstract]
- Lencioni RA, Allgaier HP, Cioni D, et al.: Small hepatocellular carcinoma in cirrhosis: randomized comparison of radio-frequency thermal ablation versus percutaneous ethanol injection. Radiology 228 (1): 235-40, 2003. [PUBMED Abstract]
- Lin SM, Lin CJ, Lin CC, et al.: Randomised controlled trial comparing percutaneous radiofrequency thermal ablation, percutaneous ethanol injection, and percutaneous acetic acid injection to treat hepatocellular carcinoma of 3 cm or less. Gut 54 (8): 1151-6, 2005. [PUBMED Abstract]
- Brunello F, Veltri A, Carucci P, et al.: Radiofrequency ablation versus ethanol injection for early hepatocellular carcinoma: A randomized controlled trial. Scand J Gastroenterol 43 (6): 727-35, 2008. [PUBMED Abstract]
- Shiina S, Teratani T, Obi S, et al.: A randomized controlled trial of radiofrequency ablation with ethanol injection for small hepatocellular carcinoma. Gastroenterology 129 (1): 122-30, 2005. [PUBMED Abstract]
- Dawson LA, Winter KA, Knox JJ, et al.: Stereotactic Body Radiotherapy vs Sorafenib Alone in Hepatocellular Carcinoma: The NRG Oncology/RTOG 1112 Phase 3 Randomized Clinical Trial. JAMA Oncol 11 (2): 136-144, 2025. [PUBMED Abstract]
- Dawson LA, Fairchild AM, Dennis K, et al.: Canadian Cancer Trials Group HE.1: A phase III study of palliative radiotherapy for symptomatic hepatocellular carcinoma and liver metastases. [Abstract] J Clin Oncol 41 (Suppl 4): A-LBA492, 2023.
Treatment of Locally Advanced or Metastatic Primary Liver Cancer
Treatment Options for Locally Advanced or Metastatic Primary Liver Cancer
Treatment options for locally advanced or metastatic primary liver cancer not amenable to surgical or locoregional interventions include:
Transarterial embolization (TAE) and transcatheter arterial chemoembolization (TACE) in patients with nonmetastatic disease
TAE is the most widely used primary treatment for hepatocellular carcinoma (HCC) not amenable to curative treatment by excision or ablation. Most of the blood supply to the normal liver parenchyma comes from the portal vein, whereas blood flow to the tumor comes mainly from the hepatic artery. Furthermore, HCC tumors are generally hypervascular compared with the surrounding normal parenchyma. The obstruction of the arterial branch(es) feeding the tumor may reduce the blood flow to the tumor and result in tumor ischemia and necrosis.
Embolization agents, such as microspheres and particles, may also be administered along with concentrated doses of chemotherapeutic agents (generally doxorubicin or cisplatin) mixed with lipiodol or other emulsifying agents during chemoembolization, arterial chemoembolization (usually via percutaneous access), and TACE. TAE-TACE is considered for patients with HCC not amenable to surgery or percutaneous ablation in the absence of extrahepatic disease.
In patients with cirrhosis, any interference with arterial blood supply may be associated with significant morbidity and is relatively contraindicated in the presence of portal hypertension, portal vein thrombosis, or clinical jaundice. In patients with liver decompensation, TAE-TACE could increase the risk of liver failure.
A number of randomized controlled trials have compared TAE and TACE with supportive care.[1] Those trials have been heterogeneous in terms of patient baseline demographics and treatment. The survival advantage of TAE-TACE over supportive care has been demonstrated by two trials.[2,3] No standardized approach for TAE has been determined (e.g., embolizing agent, chemotherapy agent and dose, and treatment schedule). However, a meta-analysis has shown that TAE-TACE improves survival more than supportive treatment.[1]
The use of drug-eluting beads (DEB) for TACE may reduce the systemic side effects of chemotherapy and may increase objective tumor response.[4–7] Only one study suggested that DEB-TACE may offer an advantage in overall survival (OS).[8]
First-line systemic therapy
Historically, sorafenib (a multikinase inhibitor) has been the standard of care for patients with advanced HCC and intact liver function (Child-Pugh class A) who were not candidates for locoregional therapy. This standard was based on the results of the SHARP trial, which showed improved OS for patients who received sorafenib compared with placebo (10.7 vs. 7.9 months; hazard ratio [HR], 0.69; P < .001). However, treatment-related adverse events may make adherence to sorafenib regimens difficult, especially in a population with concurrent liver disease. Since 2018, additional drugs and drug combinations, including atezolizumab-bevacizumab and durvalumab-tremelimumab, have resulted in improved OS when compared with sorafenib, resulting in U.S. Food and Drug Administration (FDA) approval. Other regimens have demonstrated noninferiority when compared with sorafenib, including lenvatinib (a multikinase inhibitor) and immunotherapy monotherapy. In choosing first-line therapy, survival data, response rates, bleeding risk (i.e., active varices), and the likelihood of tolerating individual therapies should be considered.
Combination immunotherapy and targeted therapy
The combination of atezolizumab (an anti–PD-L1 inhibitor) and bevacizumab (a VEGF inhibitor) has produced improved OS compared with sorafenib. The FDA approved this combination for patients with advanced HCC and Child-Pugh class A liver function. Additional combination therapies are being evaluated.
Atezolizumab and bevacizumab
Evidence (atezolizumab and bevacizumab):
- The global, open-label, phase III Imbrave150 trial (NCT03434379) included 501 patients with unresectable HCC who had not received prior systemic therapy. Patients were randomly assigned in a 2:1 ratio to receive either atezolizumab (1,200 mg intravenously [IV]) and bevacizumab (15mg/kg IV) every 3 weeks (n = 336) or sorafenib (400 mg PO bid) (n = 165). Eligibility criteria included intact liver function (Child-Pugh class A), and the study excluded patients with untreated or incompletely treated esophageal or gastric varices.[9]
- The OS was 19.2 months (95% confidence interval [CI], 17.0–23.7) in the atezolizumab-bevacizumab arm and 13.4 months (95% CI, 11.4–16.9) in the sorafenib arm (HR, 0.66; 95% CI, 0.52–0.85; P < .001).[9][Level of evidence A1]
- The objective response rates were 30% (95% CI, 25%–35%) in the atezolizumab-bevacizumab arm and 11% (95% CI, 7%–17%) in the sorafenib arm.
- In subgroup-analyses, the OS benefit was generally consistent, but with less effect in those with a nonviral etiology of HCC.
- Grade 3 or higher treatment-related adverse events occurred in 63% of patients in the atezolizumab-bevacizumab arm and 57% of patients in the sorafenib arm.[10]
Atezolizumab and cabozantinib
Evidence (atezolizumab and cabozantinib):
- The global, open-label, phase III COSMIC-312 trial (NCT03755791) included 837 patients with unresectable HCC who had not received prior systemic therapy. Patients were randomly assigned in a 2:1:1 ratio to receive either cabozantinib (40 mg PO daily) with atezolizumab (1,200 mg IV every 3 weeks), sorafenib (400 mg PO bid), or cabozantinib (60 mg daily). Eligibility criteria included intact liver function (Child-Pugh class A), and the study excluded patients with gastric or esophageal varices with active bleeding in the 6 months before enrollment.[11]
- The first primary end point explored median progression-free survival (PFS) in the first 372 patients randomly assigned to combination therapy or sorafenib. Among those patients, the median PFS was 6.8 months (99% CI, 5.6–8.3) in the cabozantinib-atezolizumab arm and 4.2 months (95% CI, 2.8–7.0) in the sorafenib arm (HR, 0.63; 99% CI, 0.44–0.91; P = .0012).[11][Level of evidence B1]
- However, at interim analysis, the OS was similar, at 15.4 months (96% CI, 13.7–17.7) for patients in the cabozantinib-atezolizumab combination arm (n = 432) and 15.5 months (12.1–not estimable) for patients in the sorafenib arm (n = 217) (HR, 0.90; 96% CI, 0.69–1.18; P = .44).
- At interim analysis, the PFS was 5.8 months (99% CI, 5.4–8.2) in the cabozantinib arm and 4.3 months (99% CI, 2.9–6.1) in the sorafenib arm (HR, 0.71; 99% CI, 0.51–1.01; P = .011).
- Objective response rates were 11% (8.1%–14.2%) in the cabozantinib-atezolizumab arm, 4% (1.6%–7.1%) in the sorafenib arm, and 6% (3.3%–10.9%) in the cabozantinib arm.
- Grade 3 or higher treatment-related adverse events occurred in 76% of patients in the cabozantinib-atezolizumab arm, 57% of patients in the sorafenib arm, and 76% of patients in the cabozantinib arm.
Combination immunotherapy alone
While single-agent immune checkpoint inhibitors have not demonstrated improved survival benefit over tyrosine kinase inhibitors (TKIs), dual immune checkpoint inhibitors have shown improved objective response rates and OS, but with increased autoimmune side effects. Optimal dosing of combination therapies is being evaluated. In 2022, based on the data below, the FDA approved the combination of a single priming dose of tremelimumab with durvalumab every 4 weeks.
Doublet immune checkpoint inhibitors
Evidence (doublet immune checkpoint inhibitors):
- The global, open-label, phase III HIMALAYA trial (NCT03298451) included 1,171 patients with unresectable HCC and Child-Pugh class A liver disease who had not received prior systemic treatment. Patients were randomly assigned in a 1:1:1 ratio to receive STRIDE (a single dose of tremelimumab 300 mg IV) with durvalumab (1,500 mg IV) every 4 weeks, durvalumab monotherapy (1,500 mg IV every 4 weeks), or sorafenib (500 mg PO bid).[12]
- The median OS was 16.43 months (95% CI, 14.16–19.58) in the combination tremelimumab-durvalumab arm and 13.77 months (95% CI, 12.25–16.13) in the sorafenib arm (HR, 0.78; 96.02% CI, 0.65–0.93; P = .0035).[12][Level of evidence A1]
- The objective response rate was 20.1% for patients who received STRIDE and 5.1% for patients who received sorafenib.
- Grade 3 or higher treatment-related adverse events occurred in 50.5% of patients who received combination tremelimumab and durvalumab and 52.4% of patients who received sorafenib.
Single-agent immune checkpoint inhibitors
Evidence (single-agent immune checkpoint inhibitors):
- The HIMALAYA trial discussed above analyzed end points for patients randomly assigned to the durvalumab monotherapy arm (n = 389) or the sorafenib monotherapy arm (n = 389).[12]
- The median OS for patients who received durvalumab monotherapy (16.56 months; 95% CI, 14.06–19.12) was noninferior to the median OS for patients who received sorafenib monotherapy (13.77 months; 95% CI, 12.25–16.13) (HR, 0.86; 95.67% CI, 0.73–1.03; noninferiority margin, 1.08).[12][Level of evidence B3]
- The objective response rate was 8.2% in the durvalumab arm and 4.9% in the sorafenib arm.
- Grade 3 or higher treatment-related adverse events occurred in 37.1% of patients who received durvalumab.
- The randomized, open-label, phase III CheckMate 459 trial (NCT02576509) included 743 patients with Child-Pugh class A liver disease and unresectable HCC who were naïve to systemic treatment. Patients were randomly assigned in a 1:1 ratio to receive either nivolumab (n = 371) or sorafenib (n = 372).[13]
- The median OS was 16.4 months (95% CI, 13.9–18.4) in the nivolumab arm and 14.7 months (95% CI, 11.9–17.2) in the sorafenib arm (HR, 0.85; 95% CI, 0.72–1.02; P = .075).[13][Level of evidence B3]
- The objective response rate was 15% (95% CI, 12%–19%) in the nivolumab arm and 7% (95% CI, 5%–10%) in the sorafenib arm.
- Grade 3 or higher treatment-related adverse events occurred in 23% of patients who received nivolumab and 49% of patients who received sorafenib.
Targeted therapy (multikinase inhibitors)
The FDA has approved two oral multikinase inhibitors, lenvatinib and sorafenib, for first-line treatment of patients with advanced HCC with well-compensated liver function who are not amenable to local therapies.
There are limited data on treatment options for patients with decompensated liver function.
Lenvatinib
Evidence (lenvatinib):
- An international, open-label, phase III, noninferiority trial (E7080-G000-304 [NCT01761266]) that included patients from 20 countries in Asia, Europe, and North America enrolled 954 patients with advanced HCC and Child-Pugh class A disease. Patients were randomly assigned in a 1:1 ratio to receive either lenvatinib (12 mg qd for body weight >60 kg or 8 mg for body weight <60 kg) or sorafenib (400 mg bid in 28-day cycles).[14] Patients with more than 50% liver involvement and portal vein invasion were excluded.
- The median OS was 13.6 months, which reached noninferiority, for patients who received lenvatinib and 12.3 months for patients who received sorafenib (HR, 0.92; 95% CI, 0.79–1.06).[14][Level of evidence B1]
- The median PFS was 7.4 months for patients who received lenvatinib and 3.7 months for patients who received sorafenib (HR, 0.66; 95% CI, 0.57–0.77).
- Treatment-related adverse events were similar between the lenvatinib arm and the sorafenib arm.
- In the lenvatinib arm, the most common side effects were hypertension (any grade, 42%), diarrhea (39%), decreased appetite (34%), and decreased weight (31%), with 11 treatment-related deaths (hepatic failure, hemorrhage, and respiratory failure).
- In the sorafenib arm, the most common side effects were palmar-plantar erythrodysesthesia (any grade, 52%), diarrhea (46%), hypertension (30%), and decreased appetite (27%), with four treatment-related deaths (hemorrhage, stroke, respiratory failure, and sudden death).
Sorafenib
Evidence (sorafenib):
- The SHARP trial (NCT00105443) randomly assigned 602 patients with advanced HCC to receive either sorafenib 400 mg twice daily or a placebo. All but 20 of the patients had a Child-Pugh class A liver disease score; 13% were women.[15]
- The study was stopped at the second planned interim analysis, after 321 deaths. The median survival was significantly longer in the sorafenib group than the placebo group (10.7 months vs. 7.9 months; HR favoring sorafenib, 0.69; 95% CI, 0.55–0.87; P < .001).
- A subsequent, similar trial was conducted in 23 centers in China, South Korea, and Taiwan. The study included 226 patients (97% with Child-Pugh class A liver function), and twice as many patients were randomly assigned to sorafenib than to placebo.[16]
- The median OS was 6.5 months for the sorafenib group versus 4.2 months for the placebo group (HR, 0.68; 95% CI, 0.50–0.93; P = .014).
Adverse events attributed to sorafenib in both of these trials included hand-foot skin reaction and diarrhea.[15,16]
Studies are also ongoing to evaluate the role of sorafenib after TACE, with chemotherapy, or in the presence of more-advanced liver disease.
Second-line systemic therapy
TKIs (regorafenib, cabozantinib, and ramucirumab) and immune checkpoint inhibitors (pembrolizumab and combination nivolumab with ipilimumab) are approved for the second-line treatment of patients with advanced HCC who have progressed while receiving sorafenib. However, the most effective second-line treatment after first-line combination atezolizumab and bevacizumab has not been determined. Physicians may consider other therapies approved in the first line (e.g., lenvatinib after atezolizumab and bevacizumab or immune checkpoint inhibitors).
Targeted therapy (multikinase inhibitors)
Regorafenib
Evidence (regorafenib):
- An international, double-blind, placebo-controlled, phase III trial (RESORCE [NCT01774344]) included patients from 21 countries in Asia, Europe, North America, South America, and Australia. The trial enrolled 573 patients with advanced HCC and Child-Pugh class A disease who had tolerated sorafenib but had disease progression. Patients were randomly assigned in a 2:1 ratio to receive either regorafenib (160 mg/day on days 1–21 of a 28-day cycle) or placebo.[17]
- The median OS was 10.6 months for patients who received regorafenib and 7.8 months for patients who received a placebo (HR, 0.63; 95% CI, 0.50–0.79).[17][Level of evidence A1]
- The median PFS was 3.1 months for patients who received regorafenib and 1.5 months for patients who received placebo.
- The most common grade 3 and 4 regorafenib-related side effects were hypertension (15%), hand-foot syndrome (13%), fatigue (9%), and diarrhea (3%).
Cabozantinib
Evidence (cabozantinib):
- An international, double-blind, placebo-controlled, phase III trial (CELESTIAL [NCT01908426]) that enrolled patients from 19 countries in Asia, Europe, North America, Australia, and New Zealand included 707 patients with advanced HCC and Child-Pugh class A disease. Patients had previously received sorafenib and progressed on at least one previous systemic therapy. Patients were randomly assigned in a 2:1 ratio to receive either cabozantinib (60 mg/day) or matching placebo. The primary end point was median OS.[18]
- The median OS was 10.2 months for patients who received cabozantinib and 8.0 months for patients who received placebo (HR, 0.76; 95% CI, 0.63–0.92, P = .005).[18][Level of evidence A1]
- The median PFS was 1.9 months for patients who received placebo and 5.2 months for patients who received cabozantinib (HR, 0.44; 95% CI, 0.36–0.52, P < .001).
- Grade 3 or 4 side effects occurred in 68% of patients who received cabozantinib compared with 37% who received placebo. Common grade 3 or 4 side effects of cabozantinib included hand-foot syndrome (17%), hypertension (16%), abnormal aspartate aminotransferase level (12%), diarrhea (11%), and fatigue (10%).
While these findings are statistically significant for OS and PFS, the absolute improvement to OS was small, toxicity (including financial toxicity) was high, and the quality-of-life data are lacking to help guide selection of regimen and who should receive treatment. These factors should all be considered and individualized for each patient.
Ramucirumab
Ramucirumab is only used in patients with alpha-fetoprotein (AFP) levels of 400 ng/mL or higher.
Evidence (ramucirumab):
- The REACH trial (NCT01140347) randomly assigned 565 patients with advanced HCC to receive either ramucirumab or placebo after first-line sorafenib. The primary end point was OS.[19][Level of evidence A1]
- The OS benefit was not statistically significant (9.2 months [95% CI, 8.0–10.6] in the ramucirumab arm and 7.6 months [95% CI, 6.0–9.3] in the placebo arm).
- An unplanned subgroup analysis showed that patients with an AFP response had improved survival compared with patients who did not.
- The REACH-2 trial (NCT02435433) included 292 patients with an Eastern Cooperative Oncology Group performance status of 0 or 1, an AFP level of at least 400 ng/mL, and Child-Pugh class A liver disease who had previously received sorafenib. Patients were randomly assigned to receive either ramucirumab or placebo.[20,21][Level of evidence A1]
- OS and PFS were improved in patients who received ramucirumab. The median OS was 8.5 months (95% CI, 7.0–10.6) for patients who received ramucirumab and 7.3 months (95% CI, 5.4–9.1) for patients who received placebo (HR, 0.710; 95% CI, 0.531–0.949; P = .0199).
- A pooled analysis of the patients in the REACH trial with an AFP greater than 400 ng/mL and the patients in REACH-2 showed improved survival regardless of Barcelona Clinic Liver Cancer (BCLC) stage.[22][Level of evidence C1]
- Among patients with BCLC stage B disease, the median OS was 13.7 months for the ramucirumab group and 8.2 months for the placebo group (HR, 0.43; 95% CI, 0.23–0.83). Among patients with BCLC stage C disease, the median OS was 7.7 months for the ramucirumab group and 4.8 months for the placebo group (HR, 0.72; 95% CI, 0.59–0.89).
Combination immunotherapy and immunotherapy alone
Pembrolizumab
Evidence (pembrolizumab):
- In an international, phase II, open-label, single-arm study (KEYNOTE-224 [NCT02702414]), 104 patients with BCLC stage B or C disease were enrolled across Europe, North America, and Japan. Patients had advanced HCC refractory to, or intolerant of, sorafenib and received pembrolizumab (200 mg IV every 3 weeks).[23]
- The phase III KEYNOTE-394 study (NCT03062358) included 453 patients in Asia with advanced HCC previously treated with sorafenib or oxaliplatin-based chemotherapy. Patients were randomly assigned in a 2:1 ratio to receive either pembrolizumab (200 mg IV) or placebo every 3 weeks for up to 35 cycles.[25]
- The OS was 14.6 months (95% CI, 12.6–18.0) in the pembrolizumab arm and 13.0 months (95% CI, 10.5–15.1) in the placebo arm (HR, 0.79; 95% CI, 0.63–0.99; P = .0180). Notably, the 24-month OS rate was 34.3% in the pembrolizumab arm and 24.9% in the placebo arm.[25][Level of evidence A1]
- Grade 3 or higher treatment-related adverse events occurred in 14.4% of patients in the pembrolizumab arm and 5.9% of patients in the placebo arm.
Based on these data, the FDA granted accelerated approval for pembrolizumab in patients with advanced HCC previously treated with sorafenib.
Nivolumab and ipilimumab
Evidence (nivolumab and ipilimumab):
- Cohort 4 of CheckMate 040 (NCT01658878), a multicenter, open-label, phase I/II study, enrolled 148 patients with advanced HCC and Child-Pugh class A liver function previously treated with sorafenib. Patients were randomly assigned in a 1:1:1 ratio to one of the following three dosages:[26]
- Arm A: Nivolumab 1 mg/kg with ipilimumab 3 mg/kg every 3 weeks for 4 doses, followed by maintenance nivolumab 240 mg every 2 weeks.
- Arm B: Nivolumab 3 mg/kg with ipilimumab 1 mg/kg every 3 weeks for 4 doses, followed by maintenance nivolumab 240 mg every 2 weeks.
- Arm C: Nivolumab 3 mg/kg every 2 weeks with ipilimumab 1 mg/kg every 6 weeks.
- The median OS was 22.8 months (95% CI, 9.4–not reached) in arm A, 12.5 months (95% CI, 7.6–16.4) in arm B, and 12.7 months (95% CI, 7.4–33.0) in arm C.[26][Level of evidence A1]
- The objective response rates were 32% (95% CI, 20%–47%) in arm A, 27% (95% CI, 15%–41%) in arm B, and 29% (95% CI, 17%–43%) in arm C.
- Grade 3 or higher treatment-related adverse events occurred in 76% of patients in arm A, 65% of patients in arm B, and 69% of patients in arm C.
Based on these data, the FDA granted accelerated approval for nivolumab (1 mg/kg IV) with ipilimumab (3 mg/kg IV every 3 weeks for 4 doses), followed by nivolumab (240 mg IV every 2 weeks) for patients with advanced HCC previously treated with sorafenib.[27]
Nivolumab
Evidence (nivolumab):
- A phase I/II, open-label, single-arm, dose-escalation and dose-expansion trial (CheckMate 040 [NCT01658878]) included 262 patients with advanced HCC and well-compensated liver function. Of those patients, 48 were enrolled in the dose-escalation phase and 214 patients were enrolled in the dose-expansion phase with nivolumab 3 mg/kg. Patients were treated with nivolumab every 2 weeks.[28] Cohorts included patients with active hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, uninfected patients with sorafenib-naïve disease, and uninfected patients with sorafenib-refractory disease.
- The total overall objective response rate in the dose-expansion phase was 20% (95% CI, 15%–26%) with three complete responses. Results were similar in untreated, refractory, and HBV/HCV-infected patients.[28][Level of evidence B4]
However, based on postmarketing requirements showing lack of confirmatory benefit, the indication for nivolumab monotherapy in the second-line setting was withdrawn in 2021.
Radiation therapy
Several phase II studies have suggested a benefit of radiation therapy in local control and OS compared with historical controls for patients with locally advanced HCC unsuitable for standard locoregional therapies.[29,30][Level of evidence C2]
Curative-intent stereotactic body radiation therapy (SBRT) improved OS in a group of patients with HCC in the NRG/RTOG 1112 study (NCT01730937), which has been presented in abstract form. Most studies have included patients with Child-Pugh class A cirrhosis. Patients with Child-Pugh class B and C cirrhosis can also be treated with liver radiation, although with a higher risk of toxicity.[31][Level of evidence B1]
Many centers deliver photon-based SBRT, while others also offer proton-based (or other heavy-ion based) radiation therapy to the liver. Based on retrospective data, proton-based radiation therapy has the potential to offer a lower dose to the normal liver and dose-escalation to the liver tumor.[32,33] Clinical trials, including NRG-GI003 (NCT03186898), are evaluating whether photon or proton therapy is superior for patients with HCC.
Palliative radiation therapy improved pain response in a randomized trial presented in abstract form. Doses commonly used included 30 Gy in ten fractions and 8 Gy in one fraction. For more information, see the Radiation therapy section in Treatment of Localized Primary Liver Cancer.[34][Level of evidence B3]
Systemic chemotherapy
There is no evidence supporting a survival benefit for patients with advanced HCC receiving systemic cytotoxic chemotherapy when compared with no treatment or best supportive care.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Llovet JM, Bruix J: Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology 37 (2): 429-42, 2003. [PUBMED Abstract]
- Llovet JM, Real MI, Montaña X, et al.: Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet 359 (9319): 1734-9, 2002. [PUBMED Abstract]
- Lo CM, Ngan H, Tso WK, et al.: Randomized controlled trial of transarterial lipiodol chemoembolization for unresectable hepatocellular carcinoma. Hepatology 35 (5): 1164-71, 2002. [PUBMED Abstract]
- Malagari K, Pomoni M, Kelekis A, et al.: Prospective randomized comparison of chemoembolization with doxorubicin-eluting beads and bland embolization with BeadBlock for hepatocellular carcinoma. Cardiovasc Intervent Radiol 33 (3): 541-51, 2010. [PUBMED Abstract]
- Varela M, Real MI, Burrel M, et al.: Chemoembolization of hepatocellular carcinoma with drug eluting beads: efficacy and doxorubicin pharmacokinetics. J Hepatol 46 (3): 474-81, 2007. [PUBMED Abstract]
- Poon RT, Tso WK, Pang RW, et al.: A phase I/II trial of chemoembolization for hepatocellular carcinoma using a novel intra-arterial drug-eluting bead. Clin Gastroenterol Hepatol 5 (9): 1100-8, 2007. [PUBMED Abstract]
- Lammer J, Malagari K, Vogl T, et al.: Prospective randomized study of doxorubicin-eluting-bead embolization in the treatment of hepatocellular carcinoma: results of the PRECISION V study. Cardiovasc Intervent Radiol 33 (1): 41-52, 2010. [PUBMED Abstract]
- Dhanasekaran R, Kooby DA, Staley CA, et al.: Comparison of conventional transarterial chemoembolization (TACE) and chemoembolization with doxorubicin drug eluting beads (DEB) for unresectable hepatocelluar carcinoma (HCC). J Surg Oncol 101 (6): 476-80, 2010. [PUBMED Abstract]
- Finn RS, Qin S, Ikeda M, et al.: Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 382 (20): 1894-1905, 2020. [PUBMED Abstract]
- Cheng AL, Qin S, Ikeda M, et al.: Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol 76 (4): 862-873, 2022. [PUBMED Abstract]
- Kelley RK, Rimassa L, Cheng AL, et al.: Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 23 (8): 995-1008, 2022. [PUBMED Abstract]
- Abou-Alfa GK, Lau G, Kudo M, et al.: Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid 1 (8): 2022. Available online. Last accessed March 26, 2025.
- Yau T, Park JW, Finn RS, et al.: Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol 23 (1): 77-90, 2022. [PUBMED Abstract]
- Kudo M, Finn RS, Qin S, et al.: Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391 (10126): 1163-1173, 2018. [PUBMED Abstract]
- Llovet JM, Ricci S, Mazzaferro V, et al.: Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359 (4): 378-90, 2008. [PUBMED Abstract]
- Cheng AL, Kang YK, Chen Z, et al.: Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 10 (1): 25-34, 2009. [PUBMED Abstract]
- Bruix J, Qin S, Merle P, et al.: Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 389 (10064): 56-66, 2017. [PUBMED Abstract]
- Abou-Alfa GK, Meyer T, Cheng AL, et al.: Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med 379 (1): 54-63, 2018. [PUBMED Abstract]
- Zhu AX, Park JO, Ryoo BY, et al.: Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol 16 (7): 859-70, 2015. [PUBMED Abstract]
- Chau I, Park JO, Ryoo BY, et al.: Alpha-fetoprotein kinetics in patients with hepatocellular carcinoma receiving ramucirumab or placebo: an analysis of the phase 3 REACH study. Br J Cancer 119 (1): 19-26, 2018. [PUBMED Abstract]
- Zhu AX, Kang YK, Yen CJ, et al.: Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 20 (2): 282-296, 2019. [PUBMED Abstract]
- Kudo M, Finn RS, Morimoto M, et al.: Ramucirumab for Patients with Intermediate-Stage Hepatocellular Carcinoma and Elevated Alpha-Fetoprotein: Pooled Results from Two Phase 3 Studies (REACH and REACH-2). Liver Cancer 10 (5): 451-460, 2021. [PUBMED Abstract]
- Zhu AX, Finn RS, Edeline J, et al.: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol 19 (7): 940-952, 2018. [PUBMED Abstract]
- Kudo M, Finn RS, Edeline J, et al.: Updated efficacy and safety of KEYNOTE-224: a phase II study of pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. Eur J Cancer 167: 1-12, 2022. [PUBMED Abstract]
- Qin S, Chen Z, Fang W, et al.: Pembrolizumab Versus Placebo as Second-Line Therapy in Patients From Asia With Advanced Hepatocellular Carcinoma: A Randomized, Double-Blind, Phase III Trial. J Clin Oncol 41 (7): 1434-1443, 2023. [PUBMED Abstract]
- Yau T, Kang YK, Kim TY, et al.: Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol 6 (11): e204564, 2020. [PUBMED Abstract]
- Saung MT, Pelosof L, Casak S, et al.: FDA Approval Summary: Nivolumab Plus Ipilimumab for the Treatment of Patients with Hepatocellular Carcinoma Previously Treated with Sorafenib. Oncologist 26 (9): 797-806, 2021. [PUBMED Abstract]
- El-Khoueiry AB, Sangro B, Yau T, et al.: Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389 (10088): 2492-2502, 2017. [PUBMED Abstract]
- Bujold A, Massey CA, Kim JJ, et al.: Sequential phase I and II trials of stereotactic body radiotherapy for locally advanced hepatocellular carcinoma. J Clin Oncol 31 (13): 1631-9, 2013. [PUBMED Abstract]
- Kawashima M, Furuse J, Nishio T, et al.: Phase II study of radiotherapy employing proton beam for hepatocellular carcinoma. J Clin Oncol 23 (9): 1839-46, 2005. [PUBMED Abstract]
- Dawson LA, Winter KA, Knox JJ, et al.: NRG/RTOG 1112: Randomized phase III study of sorafenib vs. stereotactic body radiation therapy (SBRT) followed by sorafenib in hepatocellular carcinoma (HCC). [Abstract] J Clin Oncol 41 (Suppl 4): A-489, 2023.
- Sugahara S, Oshiro Y, Nakayama H, et al.: Proton beam therapy for large hepatocellular carcinoma. Int J Radiat Oncol Biol Phys 76 (2): 460-6, 2010. [PUBMED Abstract]
- Hong TS, Wo JY, Yeap BY, et al.: Multi-Institutional Phase II Study of High-Dose Hypofractionated Proton Beam Therapy in Patients With Localized, Unresectable Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. J Clin Oncol 34 (5): 460-8, 2016. [PUBMED Abstract]
- Dawson LA, Fairchild AM, Dennis K, et al.: Canadian Cancer Trials Group HE.1: A phase III study of palliative radiotherapy for symptomatic hepatocellular carcinoma and liver metastases. [Abstract] J Clin Oncol 41 (Suppl 4): A-LBA492, 2023.
Treatment of Recurrent Primary Liver Cancer
Treatment Options for Recurrent Primary Liver Cancer
Intrahepatic recurrence is the most common pattern of failure after curative treatment.[1] Intrahepatic recurrence of hepatocellular carcinoma (HCC) may be the result of either intrahepatic metastasis or metachronous de novo tumor. Theoretically, intrahepatic metastasis may be associated with less favorable outcomes because it is most likely the result of concurrent hematogenous metastases. However, in clinical practice, the two causes of recurrence cannot be differentiated.
Treatment options for patients with recurrent primary liver cancer with liver-limited disease without vascular involvement include:
- Liver transplant.
- Surgical resection.
- Ablation.
- Radiation therapy.
Evidence (curative radiation therapy):
- A randomized controlled trial (NCT04047173) included 166 patients with recurrent HCC (after prior resection or ablation). Patients had a Karnofsky performance status score of at least 90, Child-Pugh class A cirrhosis, and a single HCC (measuring ≤5 cm). Patients were randomly assigned to receive either stereotactic body radiation therapy (SBRT) or radiofrequency ablation (RFA). The primary end point was local progression-free survival (PFS).[2]
- The local PFS rate was better with SBRT than RFA (hazard ratio [HR], 0.45, 95% confidence interval [CI], 0.24–0.87; P = .04). The 2-year local PFS rates were 92.7% (95% CI, 87.3%–98.5%) with SBRT and 75.8% (95% CI, 67.2%–85.7%) with RFA.[2][Level of evidence B1]
- There was no statistically significant difference in the 2-year OS rate between the two groups, at 97.6% (95% CI, 94.3%–100.0%) for SBRT and 93.9% (95% CI, 88.9%–99.2%) for RFA (HR, 0.91; 95% CI, 0.37–2.22; P = .830).
- The rate of adverse events was not different between the two groups.
Treatment options for patients with recurrent primary liver cancer with extrahepatic disease or vascular involvement include:
- Palliative therapy (transcatheter arterial chemoembolization [TACE] and systemic therapy).
Regarding primary HCC, the treatment strategy for recurrent intrahepatic HCC is determined by the function of the liver and the macroscopic tumor features (e.g., number of lesions, site of recurrence, and invasion of major vessels). Using the same selection criteria that are used for primary HCC, either curative (i.e., salvage liver transplant, surgical resection, and ablation) or palliative treatments (e.g., TACE and sorafenib) can be offered for recurrent HCC.
Evidence (salvage liver transplant, resection, and ablation):
- In a retrospective study of 183 patients with intrahepatic recurrence, only 87 of the patients could be treated with curative intent (transplant, resection, and ablation).[3][Level of evidence A2]
- The 5-year tumor-free survival rate was 57.9% for liver transplant, 49.3% for resection, and 10.6% for radiofrequency ablation. Subgroup analysis showed that transplant and resection led to comparable survival and that both treatments led to significantly better outcomes than did ablation (P < .001). However, selection bias was a major pitfall of this retrospective study.
- Other than the use of ablation for secondary treatment, risk factors for shorter disease-free survival were identified as alpha-fetoprotein blood levels above 400 ng/mL and recurrence within 1 year of treatment (47.5% vs. 6.7% at 5 years, P < .001).
Other studies have also suggested that most of the recurrences that appear early during follow-up are caused by tumor dissemination and have a more aggressive biological pattern than primary tumors.[4,5]
Clinical trials are appropriate and can be offered to patients with recurrent HCC whenever possible.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Fan ST, Poon RT, Yeung C, et al.: Outcome after partial hepatectomy for hepatocellular cancer within the Milan criteria. Br J Surg 98 (9): 1292-300, 2011. [PUBMED Abstract]
- Xi M, Yang Z, Hu L, et al.: Radiofrequency Ablation Versus Stereotactic Body Radiotherapy for Recurrent Small Hepatocellular Carcinoma: A Randomized, Open-Label, Controlled Trial. J Clin Oncol 43 (9): 1073-1082, 2025. [PUBMED Abstract]
- Chan AC, Chan SC, Chok KS, et al.: Treatment strategy for recurrent hepatocellular carcinoma: salvage transplantation, repeated resection, or radiofrequency ablation? Liver Transpl 19 (4): 411-9, 2013. [PUBMED Abstract]
- Minagawa M, Makuuchi M, Takayama T, et al.: Selection criteria for repeat hepatectomy in patients with recurrent hepatocellular carcinoma. Ann Surg 238 (5): 703-10, 2003. [PUBMED Abstract]
- Chen YJ, Yeh SH, Chen JT, et al.: Chromosomal changes and clonality relationship between primary and recurrent hepatocellular carcinoma. Gastroenterology 119 (2): 431-40, 2000. [PUBMED Abstract]
Latest Updates to This Summary (04/17/2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment Option Overview for Primary Liver Cancer
Revised Table 5 to include radiation therapy as a treatment option for patients with recurrent primary liver cancer with liver-limited disease without vascular involvement.
Treatment of Localized Primary Liver Cancer
Revised text about a phase III study that evaluated sorafenib alone or stereotactic body radiation therapy (SBRT) followed by sorafenib in patients with hepatocellular carcinoma (HCC) to state that the median overall survival results were not statistically significant (cited Dawson et al. as reference 16).
Treatment of Recurrent Primary Liver Cancer
Added radiation therapy as a treatment option for patients with recurrent primary liver cancer with liver-limited disease without vascular involvement.
Added text about a randomized controlled trial that included 166 patients with recurrent HCC. Patients had a Karnofsky performance status score of at least 90, Child-Pugh class A cirrhosis, and a single HCC. Patients were randomly assigned to receive either SBRT or radiofrequency ablation. The primary end point was local progression-free survival (cited Xi et al. as reference 2 and level of evidence B1).
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of adult primary liver cancer. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Primary Liver Cancer Treatment are:
- Amit Chowdhry, MD, PhD (University of Rochester Medical Center)
- Leon Pappas, MD, PhD (Massachusetts General Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Primary Liver Cancer Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/liver/hp/adult-liver-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389465]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
, each corresponding to a specific section of the liver. The boundaries of each section are separated by the right hepatic vein, middle hepatic vein, and left hepatic vein. The vena cava and portal vein are also shown.")




.")
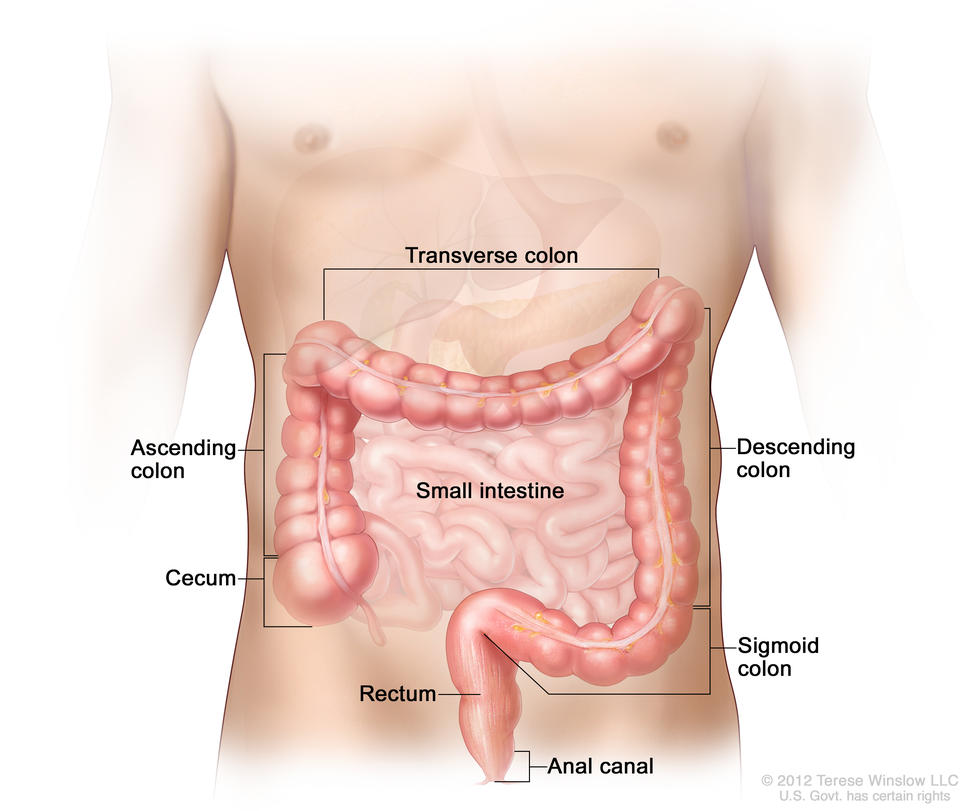
 system anatomy; drawing shows the esophagus, liver, stomach, colon, small intestine, rectum, and anus.")










 inside the colon. Inset shows photo of a pedunculated polyp.")
 kit; shows card, applicator, and return envelope.")
; drawing shows a FIT kit, which includes the package insert, the collection paper, and a collection tube with a small brush inside of it. Also shown are the biohazard bag, the return envelope, and a paper with information about colorectal cancer and colorectal cancer screening.")



