
Researchers are testing certain targeted therapies for some types of endometrial cancer.
Credit: iStock
NCI-funded researchers are working to advance our understanding of how to prevent, detect, and treat endometrial cancer, which is a type of uterine cancer. The other type, uterine sarcoma, is much less common and can be more aggressive and harder to treat.
There are two main subtypes of endometrial cancers: endometrioid and non-endometrioid. Both occur in the inner lining of the uterus, but they look different under a microscope.
- Endometrioid tumors are more common (they make up 75% to 80% of uterine cancers), are typically diagnosed at an early stage, and may have a favorable prognosis.
- Non-endometrioid tumors (including serous, clear cell, carcinosarcoma, and other, rarer types of endometrial cancer) are often more aggressive and have a poor prognosis.
This page highlights some of the latest research in endometrial cancer, NCI-supported programs that are fueling progress, and research findings from recent studies.
Early Detection of Endometrial Cancer
There is no standard screening test for endometrial cancer. Researchers are exploring a variety of ways to detect endometrial cancer before symptoms develop. This includes studying genetic risk factors that increase the risk of endometrial and other cancers.
Abnormal bleeding: Early-stage endometrial cancer and even atypical hyperplasia of the endometrium (which is not cancer but can become cancer) can cause vaginal bleeding in postmenopausal women. Although bleeding can have many causes, research shows that most postmenopausal women with endometrial cancer had abnormal vaginal bleeding before diagnosis. This confirms the value of follow-up testing in women who have this symptom.
New biomarkers: Scientists are looking at potential biomarkers to further improve diagnosis of early endometrial cancer. A biomarker is a molecule found in blood or other tissues that is a sign of a condition or disease. Research has shown that it’s possible to detect endometrial cancer biomarkers from minimally invasive, lower genital tract samples.
In the DETECT Study, for example, researchers from NCI’s Division of Cancer Epidemiology and Genetics (DCEG) are studying ways to detect endometrial cancer in samples collected using vaginal tampons. Scientists are comparing biomarkers in both tissue and tampon samples collected from women who are having a hysterectomy for endometrial cancer, and from women having a hysterectomy for an unrelated benign condition. Researchers hope to find biomarkers that may eventually lead to noninvasive early detection approaches. This study is also designed to reach a racially diverse group of women.
Researchers funded by NCI’s Early Detection Research Network (EDRN), a network of institutions developing biomarkers to detect cancer in its early stages, designed a test called PapSEEK that analyzes cells from the lining of the uterus. In a research study, the test identified cancer-related DNA alterations in most women with known endometrial cancer, but also in a few women without the disease.
More studies of PapSEEK are needed before the test will be ready for use in patient care.
Familial genetic risk: Lynch syndrome is an inherited DNA repair disorder in which people have a higher-than-normal risk of developing certain cancers, including endometrial cancer, colon cancer, and, less frequently, ovarian cancer. About 5% of endometrial cancers are caused by Lynch syndrome. It is recommended that all women diagnosed with endometrial cancer be tested for this disorder. This will aid in treatment decisions and also help with prevention and screening of other cancers in the patient and their blood relatives.
Advances in Endometrial Cancer Treatment
Surgery is the standard treatment for early-stage endometrial cancer. Additional treatment, depending on the stage of disease and other factors, may include radiation with or without chemotherapy, hormone therapy, immunotherapy, and some targeted therapies. Several new treatments for advanced disease have become available. (For a complete list of all currently approved drugs, see Drugs Approved for Endometrial Cancer.)
Molecular Subtypes
One area that is changing practice is determining the molecular subtypes of cancers and deciding treatment according to type. Funded by the Cancer Genome Atlas Program, researchers have found that there are four molecular subtypes of endometrial cancer. These subtypes differ in how likely it is that the cancer will come back after treatment.
Doctors are now using these subtypes to help choose the best treatments for certain patients with endometrial cancer. Molecular analysis of endometrial cancers is now recommended for all newly diagnosed patients and can be used to guide treatment decisions in selected subtypes. This includes intensifying treatment where needed, or reducing the intensity of treatment if it’s shown to be safe and equally effective.
Immunotherapy
Immunotherapies help the immune system to better fight cancer. Immune checkpoint inhibitors, a type of immunotherapy, have shown promise in treating certain forms of endometrial cancer.
These drugs are especially useful in tumors that have defects in a specific DNA repair process, called mismatch repair. Tumors with mismatch repair deficiency (dMMR) develop a large number of DNA mutations, a condition called high microsatellite instability (MSI-H). Such tumors are particularly vulnerable to treatment with immunotherapy alone or immunotherapy in combination with other therapies.
Endometrial cancers that develop in people with Lynch syndrome are dMMR/MSI-H. In addition, around one-third of people with endometrial cancer that is not due to an inherited defect in DNA repair also have dMMR/MSI-H cancers.
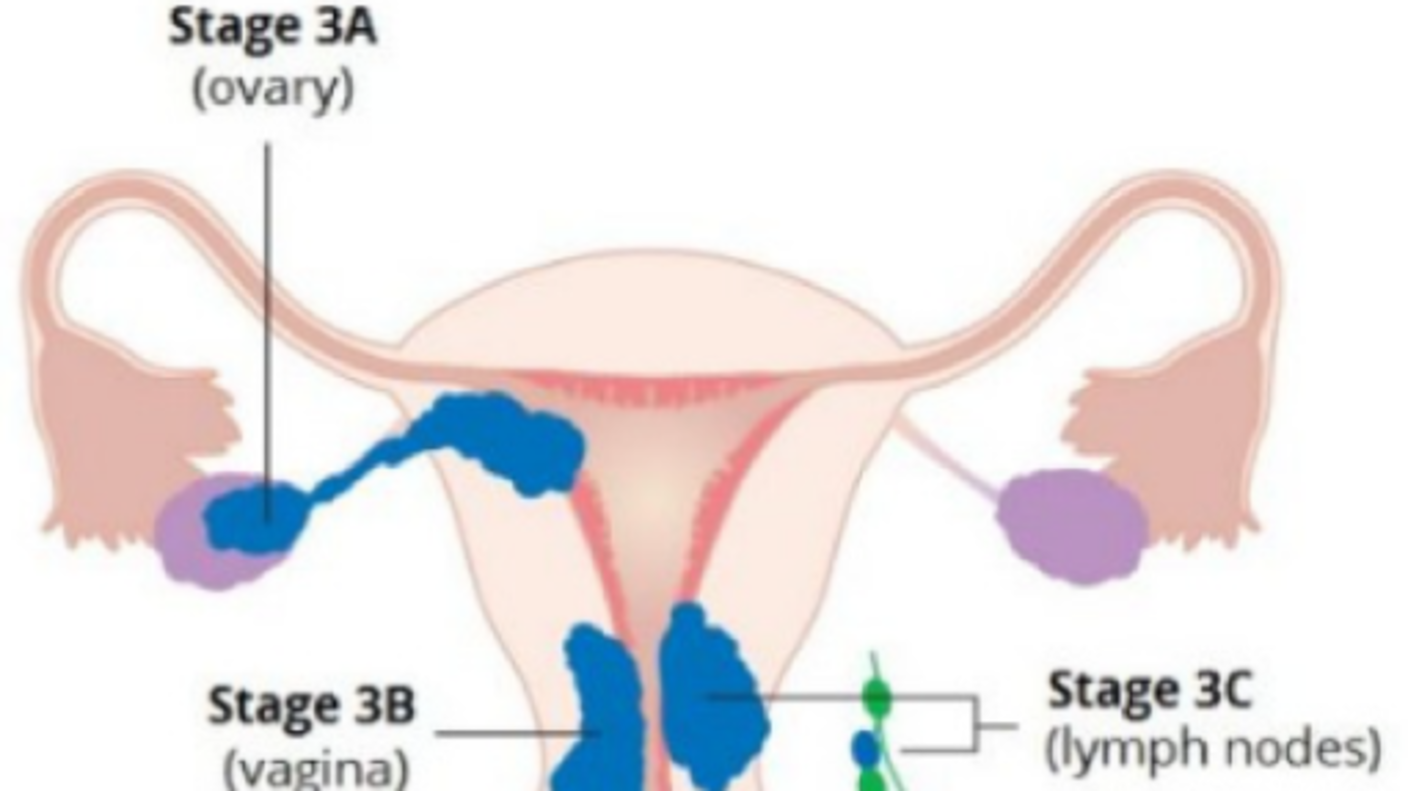
The immune checkpoint inhibitor pembrolizumab (Keytruda) has been approved for treating patients with advanced endometrial cancer that is dMMR or MSI-H, cannot be removed surgically, and has gotten worse after other treatments. A different immune checkpoint inhibitor, dostarlimab, is also used for advanced endometrial cancer that is dMMR and is not responding to chemotherapy.
When combined with chemotherapy, both drugs have been shown to extend the time until disease recurs. This applies to patients with newly diagnosed advanced stage endometrial cancer or those with a first recurrence after radiation therapy.
The chemotherapy/dostarlimab combination was approved for use in patients with dMMR cancers. It is expected that the NCI-supported trial of chemotherapy/pembrolizumab will be approved for dMMR patients. The pembrolizumab study suggests there may also be benefit of the combination for patients who do not have dMMR cancers, but conclusions are pending.
Other advances include:
- An NCI-supported study is testing whether combining the drugs nivolumab and ipilimumab is better than nivolumab alone in shrinking tumors in patients with recurrent endometrial carcinoma that has progressed after earlier treatment with an immune checkpoint inhibitor. The combination of these two drugs has been found to shrink or stabilize other dMMR cancers, but this has not been studied in endometrial cancer until now.
- Pembrolizumab has also been approved to be used together with the targeted therapy lenvatinib (Lenvima) for some patients with advanced endometrial cancer that is not MSI-H or dMMR and has gotten worse after other treatments. A 2022 clinical trial showed that combining the two drugs led to longer progression-free survival and overall survival among patients than using chemotherapy.
- Researchers are also examining the role of adding pembrolizumab to standard radiation therapy for early-stage endometrial cancer that is MSI-H or dMMR.
Targeted Therapy
Targeted therapies are drugs or other substances that interfere with specific molecules, or targets, to block the growth and spread of cancer with less harm to normal cells.
Several targeted therapies are being studied for treating advanced endometrial cancer. Some examples include:
- One NCI-supported trial is studying how well the drugs olaparib (Lynparza) and cediranib maleate (Recentin) work in treating patients with endometrial cancer that has come back, does not respond to treatment, or has spread elsewhere in the body. These drugs may stop the growth of tumor cells by blocking some of the enzymes needed for cell growth. This phase 2 clinical trial is now testing three additional combinations of targeted therapies.
- A new trial is testing the combination of olaparib with the chemotherapy drug temozolomide in people with a type of uterine sarcoma called leiomyosarcoma. The hope is that using both drugs together will work better to treat the disease than giving either drug alone.
- Another trial is testing whether adding certain targeted therapies to chemotherapy will shrink tumors in patients with one of two rare types of endometrial cancer that have excess amounts of a protein called HER2 (also called HER2 positive cancer). The treatment will target the HER2 protein and will be given in a new form, a subcutaneous shot (under the skin), rather than patients having another IV infusion.
Treatment Combinations
Radiation therapy and cisplatin: An NCI randomized phase 2 trial is comparing the combination of radiation therapy and cisplatin with radiation therapy alone in treating patients with endometrial cancer that has come back. The trial is now closed and researchers are analyzing the results.
Surgery and chemotherapy versus surgery and chemoradiation: An NCI-funded study found that, among women with locally advanced endometrial cancer, those who received radiation in addition to chemotherapy (chemoradiation) after surgery had the same rate of cancer recurrence as those who received chemotherapy without radiation. More research is needed to determine whether specific groups of patients would benefit from radiation.
Rising Endometrial Cancer Rates and Disparities
Unlike most other cancers in the United States, endometrial cancer has increased in both incidence and death rates in recent years. These changes reflect increases in aggressive (non-endometrioid) subtypes of uterine cancer, with rates of endometrioid subtypes having remained fairly stable.
Recent studies have shown that these increases are seen in all racial and ethnic groups. However, a 2019 study from NCI showed that Black women have the highest incidence rates and poorer survival than women in other racial and ethnic groups. In a 2022 NCI study, Black women had more than twice the rate of deaths from uterine cancer overall compared with other racial and ethnic groups. This may be due to a higher frequency of the serous subtype of endometrial cancer in Black women, but scientists are studying why this might be the case.
The reasons for the increases in non-endometrioid subtypes and the disparities across groups are not clear, but NCI-funded studies are seeking to understand their origin. For example:
- In addition to studying biomarkers in tampon specimens, the aforementioned DETECT study has expanded their aims to investigate possible sources of these disparities, such as differences in risk factors, in molecular markers and in care delays.
- As part of NCI’s Cancer Moonshot Program, researchers at Ohio State University will examine the genomics of 350 Black and 350 white women with higher risk endometrial cancers. Scientists hope to get a better understanding of the underlying biology of these tumors in order to better personalize treatment.
- The Social Interventions for Support During Treatment for Patients with Endometrial Cancer (SISTER Study) will compare whether weekly support groups led by peer supporters, 1-on-1 peer support check-ins, or enhanced usual care work better to support Black patients with endometrial cancer during treatment. Researchers hope to see if social interventions can provide support and improve the well-being and quality of life of patients with endometrial cancer.
- In the NIH-funded, Multilevel determinants of racial disparities in receipt of guideline-concordant endometrial cancer treatment, researchers at Ohio State University will analyze data from NCI’s Surveillance, Epidemiology, and End Results (SEER) Medicare database and conduct interviews with Black women with endometrial cancer. They hope to find out what causes the differences in how this group gets treated compared to the recommended guidelines for treatment.
- The Carolina Endometrial Cancer Study seeks to address this gap by analyzing endometrial tumors to identify genetic details and guide treatment strategies. Women from across the state of North Carolina are being recruited, with a goal of half the participants being Black.
NCI-Supported Research Programs
Many NCI-funded researchers at the NIH campus, and across the United States and the world, are seeking ways to address uterine cancer more effectively. Some research is basic, exploring questions as diverse as the biological underpinnings of cancer and the social factors that affect cancer risk. And some is more clinical, seeking to translate this basic information into improving patient outcomes.
- The Endometrial Specialized Programs of Research Excellence (SPOREs) promotes collaborative translational cancer research. This group works to improve prevention and treatment approaches, along with molecular diagnostics, in the clinical setting to help patients with endometrial cancer.
- NCI”s Division of Cancer Prevention (DCP) is addressing rising endometrial cancer rates by supporting gynecologic cancer prevention research and developing concepts for future studies.
- Approaches to Identify and Care for Individuals with Inherited Cancer Syndromes seeks the best approaches to identify those with an inherited cancer syndrome and provide appropriate follow-up care.
- The NCI-funded Colon Cancer Family Registry has established an international cohort of thousands of colorectal cancer patients, their relatives, and other individuals at increased risk of colorectal and other cancers, including endometrial cancer. More than 10,000 families from the United States, Canada, Australia, and New Zealand have been registered. The database includes more than 2,000 individuals with Lynch syndrome from 781 families.
- The Epidemiology of Endometrial Cancer Consortium (E2C2) is an NCI-supported consortium studying the causes and origins of this cancer through collaboration among investigators. The goal of E2C2 is to combine data across studies to better understand endometrial cancer.
Clinical Trials for Uterine Cancer
NCI funds and oversees both early- and late-phase clinical trials to develop new treatments and improve patient care. Trials are available for the treatment of both endometrial cancer and uterine sarcoma.
Endometrial Cancer Research Results
The following are some of our latest news articles on endometrial cancer research:
- More Immunotherapy Options Approved for Treating Endometrial Cancer
- Immunotherapy’s Role in Treating Endometrial Cancer Expected to Grow
- Uterine Cancer Death Rates Rising, Highest Among Black Women in the United States
- Trastuzumab May Improve Survival in Women with Rare Endometrial Cancer
- Women Experience More Side Effects from Pelvic Radiation than Realized
- Can Some Women Treated for Endometrial Cancer Forgo Radiation after Surgery?
View the full list of Uterine Cancer Research Results and Study Updates.
, endometrium (inner lining of the uterus), ovaries, fallopian tubes, cervix, and vagina.")
. Three-panel drawing showing a side view of the female reproductive anatomy during a D and C procedure. The first panel shows a speculum widening the opening of the vagina. The cervix, uterus with abnormal tissue, bladder, and rectum are also shown; an inset shows the lower half of a woman covered by a drape on an exam table with her legs apart and her feet in stirrups. The middle panel shows the uterus and a dilator inserted through the vagina into the cervix. The third panel shows a curette scraping out abnormal tissue from the uterus; an inset shows a close up of the curette with the abnormal tissue in it.")
 is shown inserted into the vagina. The bladder, uterus, right fallopian tube, and right ovary are also shown. The inset shows the diagnostic sonographer (a person trained to perform ultrasound procedures) examining a woman on a table, and a computer screen shows an image of the patient’s internal tissues.")









 scan; drawing shows a child lying on a table that slides into the MRI machine, which takes a series of detailed pictures of areas inside the body.")
 scan; drawing shows a child lying on a table that slides through the CT scanner, which takes a series of detailed x-ray pictures of areas inside the body.")
 scan; drawing shows a child lying on table that slides through the PET scanner.")



 to antigen and MHC proteins on the antigen-presenting cell (APC) and the binding of CD28 on the T cell to B7-1/B7-2 on the APC. It also shows the binding of B7-1/B7-2 to CTLA-4 on the T cell, which keeps the T cells in the inactive state. The panel on the right shows immune checkpoint inhibitor (anti-CTLA antibody) blocking the binding of B7-1/B7-2 to CTLA-4, which allows the T cells to be active and to kill tumor cells.")
 to PD-1 (on the T cell), which keeps T cells from killing tumor cells in the body. Also shown are a tumor cell antigen and T cell receptor. The panel on the right shows immune checkpoint inhibitors (anti-PD-L1 and anti-PD-1) blocking the binding of PD-L1 to PD-1, which allows the T cells to kill tumor cells.")