
Hormone Therapy for Breast Cancer
What are hormones and hormone receptors?
Hormones are substances that function as chemical messengers in the body. They affect the actions of cells and tissues at various locations in the body, often reaching their targets through the bloodstream.
The hormones estrogen and progesterone are produced by the ovaries in premenopausal women and by some other tissues, including fat and skin, in both premenopausal and postmenopausal women and in men. Estrogen promotes the development and maintenance of female sex characteristics and the growth of long bones. Progesterone plays a role in the menstrual cycle and pregnancy.
Estrogen and progesterone also promote the growth of some breast cancers, which are called hormone-sensitive (or hormone-dependent) breast cancers. Hormone-sensitive breast cancer cells contain proteins called hormone receptors (estrogen receptors, or ERs, and progesterone receptors, or PRs) that become activated when hormones bind to them. The activated receptors cause changes in the expression of specific genes, which can stimulate cell growth.
To determine whether breast cancer cells contain hormone receptors, doctors test samples of tumor tissue that have been removed by surgery. If the tumor cells contain estrogen receptors, the cancer is called estrogen receptor positive (ER positive), estrogen sensitive, or estrogen responsive. Similarly, if the tumor cells contain progesterone receptors, the cancer is called progesterone receptor positive (PR or PgR positive). Breast tumors that contain estrogen and/or progesterone receptors are sometimes called hormone receptor positive (HR positive). Most ER-positive breast cancers are also PR positive.
Breast cancers that lack ERs are called ER negative, and if they lack both ER and PR they may be called HR negative.
Approximately 67%–80% of breast cancers in women are ER positive (1, 2). Approximately 90% of breast cancers in men are ER positive and approximately 80% are PR positive (3).
What is hormone therapy?
Hormone therapy (also called hormonal therapy, hormone treatment, or endocrine therapy) slows or stops the growth of hormone-sensitive tumors by blocking the body’s ability to produce hormones or by interfering with effects of hormones on breast cancer cells. Tumors that are hormone insensitive do not have hormone receptors and do not respond to hormone therapy.
Hormone therapy for breast cancer should not be confused with menopausal hormone therapy (MHT)—treatment with estrogen alone or in combination with progesterone to help relieve symptoms of menopause. These two types of therapy produce opposite effects: hormone therapy for breast cancer blocks the growth of HR-positive breast cancer, whereas MHT can stimulate the growth of HR-positive breast cancer. For this reason, when a woman taking MHT is diagnosed with HR-positive breast cancer she is usually asked to stop that therapy.
What types of hormone therapy are used for breast cancer?
Several strategies are used to treat hormone-sensitive breast cancer:
Blocking ovarian function: Because the ovaries are the main source of estrogen in premenopausal women, estrogen levels in these women can be reduced by eliminating or suppressing ovarian function. Blocking ovarian function is called ovarian ablation.
Ovarian ablation can be done surgically in an operation to remove the ovaries (called oophorectomy) or by treatment with radiation. This type of ovarian ablation is usually permanent.
Alternatively, ovarian function can be suppressed temporarily by treatment with drugs called gonadotropin-releasing hormone (GnRH) agonists, which are also known as luteinizing hormone-releasing hormone (LHRH) agonists. By mimicking GnRH, these medicines interfere with signals that stimulate the ovaries to produce estrogen.
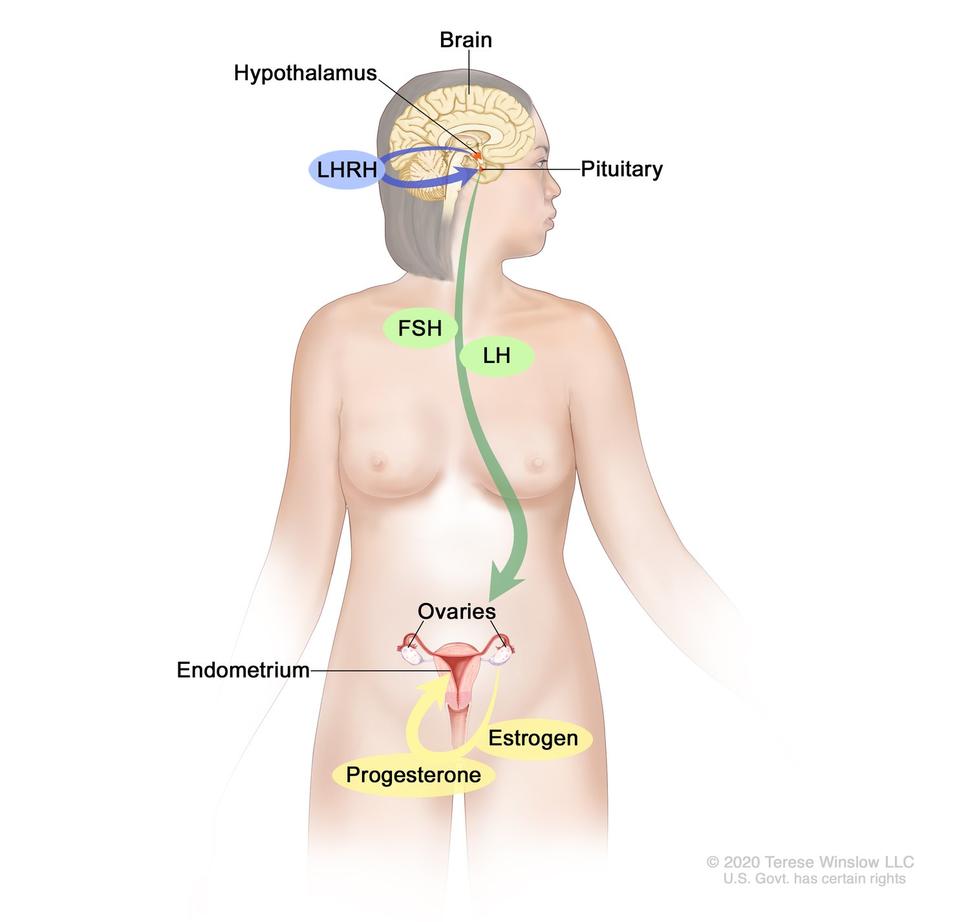
Estrogen and progesterone production in premenopausal women. Drawing shows that in premenopausal women, estrogen and progesterone production by the ovaries is regulated by luteinizing hormone (LH) and luteinizing hormone-releasing hormone (LHRH). The hypothalamus releases LHRH, which then causes the pituitary gland to make and secrete LH and follicle-stimulating hormone (FSH). LH and FSH cause the ovaries to make estrogen and progesterone, which act on the endometrium (inner lining of the uterus). (When estrogen and progesterone production reaches a certain level during the menstrual cycle, these hormones act on the hypothalamus and pituitary to turn off production of LHRH, LH, and FSH.)
Examples of ovarian suppression drugs are goserelin (Zoladex) and leuprolide (Lupron).
Blocking estrogen production: Drugs called aromatase inhibitors are used to block the activity of an enzyme called aromatase, which the body uses to make estrogen in the ovaries and in other tissues. Aromatase inhibitors are used primarily in postmenopausal women because the ovaries in premenopausal women produce too much aromatase for the inhibitors to block effectively. However, these drugs can be used in premenopausal women if they are given together with a drug that suppresses ovarian function.
Examples of aromatase inhibitors approved by the FDA are anastrozole (Arimidex) and letrozole (Femara), both of which temporarily inactivate aromatase, and exemestane (Aromasin), which permanently inactivates aromatase.
Blocking estrogen’s effects: Several types of drugs interfere with estrogen’s ability to stimulate the growth of breast cancer cells:
- Selective estrogen receptor modulators (SERMs) bind to estrogen receptors, preventing estrogen from binding. Examples of SERMs approved by the FDA for treatment of breast cancer are tamoxifen (Nolvadex) and toremifene (Fareston).
Because they bind to estrogen receptors, SERMs can potentially not only block estrogen activity (by preventing estrogen from binding to its receptor) but also mimic the effects of estrogen, depending on where they are expressed in the body. For example, tamoxifen blocks the effects of estrogen in breast tissue but acts like estrogen in the uterus and bone.
- Other antiestrogen drugs, such as fulvestrant (Faslodex), work in a somewhat different way to block estrogen’s effects. Like SERMs, fulvestrant binds to the estrogen receptor and functions as an estrogen blocker. However, unlike SERMs, fulvestrant does not mimic estrogen. For this reason, it is called a pure antiestrogen. In addition, when fulvestrant binds to the estrogen receptor, the receptor is targeted for destruction.
How is hormone therapy used to treat breast cancer?
There are three main ways that hormone therapy is used to treat hormone-sensitive breast cancer:
Adjuvant therapy for early-stage breast cancer: Tamoxifen is FDA approved for adjuvant hormone treatment of premenopausal and postmenopausal women (and men) with ER-positive early-stage breast cancer, and the aromatase inhibitors anastrozole, letrozole, and exemestane are approved for this use in postmenopausal women.
Research has shown that women who receive at least 5 years of adjuvant therapy with tamoxifen after having surgery for early-stage ER-positive breast cancer have reduced risks of breast cancer recurrence, including a new breast cancer in the other breast, and reduced risk of death at 15 years (4).
Until recently, most women who received adjuvant hormone therapy to reduce the chance of a breast cancer recurrence took tamoxifen every day for 5 years. However, with the introduction of newer hormone therapies (i.e., the aromatase inhibitors), some of which have been compared with tamoxifen in clinical trials, additional approaches to hormone therapy have become common (5–7).
For example, some women may take an aromatase inhibitor, instead of tamoxifen, every day for 5 years. Other women may receive additional treatment with an aromatase inhibitor after 5 years of tamoxifen. Finally, some women may switch to an aromatase inhibitor after 2 or 3 years of tamoxifen, for a total of 5 or more years of hormone therapy. Research has shown that for postmenopausal women who have been treated for early-stage breast cancer, adjuvant therapy with an aromatase inhibitor reduces the risk of recurrence and improves overall survival compared with adjuvant tamoxifen (8).
Some premenopausal women with early-stage ER-positive breast cancer may have ovarian suppression plus an aromatase inhibitor, which was found to have higher rates of freedom from recurrence than ovarian suppression plus tamoxifen or tamoxifen alone (9).
Men with early-stage ER-positive breast cancer who receive adjuvant therapy are usually treated first with tamoxifen. Those treated with an aromatase inhibitor usually also take a GnRH agonist.
Decisions about the type and duration of adjuvant hormone therapy are complicated and must be made on an individual basis in consultation with an oncologist.
Treatment of advanced or metastatic breast cancer: Several types of hormone therapy are approved to treat metastatic or recurrent hormone-sensitive breast cancer. Hormone therapy is also a treatment option for ER-positive breast cancer that has come back in the breast, chest wall, or nearby lymph nodes after treatment (also called a locoregional recurrence).
Two SERMs, tamoxifen and toremifene, are approved to treat metastatic breast cancer. The antiestrogen fulvestrant is approved for postmenopausal women with metastatic ER-positive breast cancer that has spread after treatment with other antiestrogens (10). Fulvestrant is also approved for postmenopausal women with HR-positive, HER2-negative locally advanced or metastatic breast cancer who have not previously been treated with hormone therapy (11). In addition, it may be used in premenopausal women who have had ovarian ablation.
The aromatase inhibitors anastrozole and letrozole are approved to be given to postmenopausal women as initial therapy for metastatic or locally advanced hormone-sensitive breast cancer (12, 13). Both of these drugs and the aromatase inhibitor exemestane are also approved to treat postmenopausal women with advanced breast cancer whose disease has worsened after treatment with tamoxifen (14). Men with advanced breast cancer who are treated with an aromatase inhibitor also receive a GnRH agonist.
Some women with advanced breast cancer are treated with a combination of hormone therapy and one of several targeted therapies:
- Palbociclib (Ibrance), is approved for use in combination with letrozole as initial therapy for the treatment of HR-positive, HER2-negative advanced or metastatic breast cancer in postmenopausal women. Palbociclib inhibits two cyclin-dependent kinases (CDK4 and CDK6) that appear to promote the growth of HR-positive breast cancer cells (15).
Palbociclib is also approved to be used in combination with fulvestrant for the treatment of postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer whose cancer has gotten worse after treatment with another hormone therapy (16).
- Abemaciclib (Verzenio), another CDK4 and CDK6 inhibitor, is approved to be used in combination with fulvestrant for postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer whose disease has progressed after treatment with hormone therapy (17).
Abemaciclib is also approved to be used alone for women and men with HR-positive, HER2-negative advanced or metastatic breast cancer whose disease got worse after treatment with hormone therapy and previous chemotherapy given for metastatic disease (18).
Abemaciclib is also approved to be used with an aromatase inhibitor as first-line hormone therapy in postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer.
- Ribociclib (Kisqali), another CDK4/6 inhibitor, is approved to be used in combination with an aromatase inhibitor in postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer that has not been treated with hormone therapy (19, 20).
Ribociclib is also approved to be used in combination with fulvestrant in postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer who have not been treated with hormone therapy or whose disease got worse during treatment with hormone therapy (21).
- Lapatinib (Tykerb) is approved to be used in combination with letrozole to treat HR-positive, HER2-positive metastatic breast cancer in postmenopausal women for whom hormone therapy is indicated. It is a small-molecule inhibitor of the HER2 and EGFR tyrosine kinases.
- Alpelisib (Piqray) is approved to treat breast cancer that is HR positive and HER2 negative and has a mutation in the PIK3CA gene. It is used with fulvestrant to treat postmenopausal women, and men, whose breast cancer is advanced or metastatic and has gotten worse during or after treatment with hormone therapy (22).
- Some women with advanced breast cancer that is HER2 and HR positive may receive hormone therapy plus trastuzumab with or without pertuzumab (23).
Neoadjuvant treatment of breast cancer: The use of hormone therapy to treat breast cancer to reduce tumor size before surgery (neoadjuvant therapy) has been studied in clinical trials (24). These trials have shown that neoadjuvant hormone therapy—in particular, with aromatase inhibitors—can be effective in reducing the size of breast tumors in postmenopausal women, but it is not yet clear how effective it is in premenopausal women.
Hormone therapy is sometimes used for the neoadjuvant treatment of HR-positive breast cancer in postmenopausal women who cannot tolerate chemotherapy or when surgery needs to be delayed.
Can hormone therapy be used to prevent breast cancer?
Yes. Most breast cancers are ER positive, and clinical trials have tested whether hormone therapy can be used to prevent breast cancer in women who are at increased risk of developing the disease.
A large NCI-sponsored randomized clinical trial called the Breast Cancer Prevention Trial found that tamoxifen, taken for 5 years, reduces the risk of developing invasive breast cancer by about 50% in postmenopausal women who were at increased risk (25). Long-term follow-up of another randomized trial, the International Breast Cancer Intervention Study I, found that 5 years of tamoxifen treatment reduces the incidence of breast cancer for at least 20 years (26). A subsequent large randomized trial, the Study of Tamoxifen and Raloxifene, which was also sponsored by NCI, found that 5 years of raloxifene (a SERM) reduces breast cancer risk in such women by about 38% (27).
As a result of these trials, both tamoxifen and raloxifene have been approved by the FDA to reduce the risk of developing breast cancer in women at high risk of the disease. Tamoxifen is approved for this use regardless of menopausal status. Raloxifene is approved for use only in postmenopausal women.
Two aromatase inhibitors—exemestane and anastrozole—have also been found to reduce the risk of breast cancer in postmenopausal women at increased risk of the disease. After 3 years of follow-up in a randomized trial, women who took exemestane were 65% less likely than those who took a placebo to develop breast cancer (28). After 7 years of follow-up in another randomized trial, women who took anastrozole were 50% less likely than those who took a placebo to develop breast cancer (29). Both exemestane and anastrozole are approved by the FDA for treatment of women with ER-positive breast cancer. Although both are also used for breast cancer prevention, neither is approved for that indication specifically.
What are the side effects of hormone therapy?
The side effects of hormone therapy depend largely on the specific drug or the type of treatment (7). The benefits and harms of taking hormone therapy should be carefully weighed for each person. A common switching strategy used for adjuvant therapy, in which patients take tamoxifen for 2 or 3 years, followed by an aromatase inhibitor for 2 or 3 years, may yield the best balance of benefits and harms of these two types of hormone therapy (30).
Hot flashes, night sweats, and vaginal dryness are common side effects of all hormone therapies. Hormone therapy also may disrupt the menstrual cycle in premenopausal women.
Less common but serious side effects of hormone therapy drugs are listed below.
Tamoxifen
- risk of blood clots, especially in the lungs and legs
- stroke
- cataracts
- endometrial cancer and uterine sarcoma
- bone loss in premenopausal women, but no increased risk of fracture
- mood swings, depression, and loss of libido
- in men: headaches, nausea, vomiting, skin rash, impotence, and loss of libido
Raloxifene
- risk of blood clots, especially in the lungs and legs
- stroke in certain subgroups
Ovarian suppression
- bone loss
- mood swings, depression, and loss of libido
Aromatase inhibitors
- risk of heart attack, angina, heart failure, and hypercholesterolemia
- bone loss
- joint pain
- mood swings and depression
Fulvestrant
- gastrointestinal symptoms, including nausea, vomiting, and constipation
- weakness and fatigue
- pain, including bone pain, back pain, musculoskeletal pain, joint pain, and in the extremities
- headache
- hot flashes
- breathing problems, including painful breathing, shortness of breath, and cough
- loss of appetite
Can other drugs interfere with hormone therapy?
Certain drugs, including several commonly prescribed antidepressants (those in the category called selective serotonin reuptake inhibitors, or SSRIs), inhibit an enzyme called CYP2D6. This enzyme plays a critical role in the body’s use of tamoxifen because CYP2D6 metabolizes, or breaks down, tamoxifen into molecules, or metabolites, that are much more active than tamoxifen itself.
The possibility that SSRIs might, by inhibiting CYP2D6, slow the metabolism of tamoxifen and reduce its effectiveness is a concern given that as many as one-fourth of breast cancer patients experience clinical depression and may be treated with SSRIs. In addition, SSRIs are sometimes used to treat hot flashes caused by hormone therapy.
Many experts suggest that patients who are taking antidepressants along with tamoxifen should discuss treatment options with their doctors, such as switching from an SSRI that is a potent inhibitor of CYP2D6, such as paroxetine hydrochloride (Paxil), to one that is a weaker inhibitor, such as sertraline (Zoloft) or citalopram (Celexa), or to an antidepressant that does not inhibit CYP2D6, such as venlafaxine (Effexor) (31). Or doctors may suggest that their postmenopausal patients take an aromatase inhibitor instead of tamoxifen.
Other medications that inhibit CYP2D6 include the following:
- quinidine, which is used to treat abnormal heart rhythms
- diphenhydramine, which is an antihistamine
- cimetidine, which is used to reduce stomach acid
People who are prescribed tamoxifen should discuss the use of all other medications with their doctors.
 scan; drawing shows a child lying on a table that slides into the MRI machine, which takes a series of detailed pictures of areas inside the body.")
 scan; drawing shows a child lying on a table that slides through the CT scanner, which takes a series of detailed x-ray pictures of areas inside the body.")
 scan; drawing shows a child lying on table that slides through the PET scanner.")


, endometrium (inner lining of the uterus), ovaries, fallopian tubes, cervix, and vagina.")
. Three-panel drawing showing a side view of the female reproductive anatomy during a D and C procedure. The first panel shows a speculum widening the opening of the vagina. The cervix, uterus with abnormal tissue, bladder, and rectum are also shown; an inset shows the lower half of a woman covered by a drape on an exam table with her legs apart and her feet in stirrups. The middle panel shows the uterus and a dilator inserted through the vagina into the cervix. The third panel shows a curette scraping out abnormal tissue from the uterus; an inset shows a close up of the curette with the abnormal tissue in it.")
 is shown inserted into the vagina. The bladder, uterus, right fallopian tube, and right ovary are also shown. The inset shows the diagnostic sonographer (a person trained to perform ultrasound procedures) examining a woman on a table, and a computer screen shows an image of the patient’s internal tissues.")










 to antigen and MHC proteins on the antigen-presenting cell (APC) and the binding of CD28 on the T cell to B7-1/B7-2 on the APC. It also shows the binding of B7-1/B7-2 to CTLA-4 on the T cell, which keeps the T cells in the inactive state. The panel on the right shows immune checkpoint inhibitor (anti-CTLA antibody) blocking the binding of B7-1/B7-2 to CTLA-4, which allows the T cells to be active and to kill tumor cells.")
 to PD-1 (on the T cell), which keeps T cells from killing tumor cells in the body. Also shown are a tumor cell antigen and T cell receptor. The panel on the right shows immune checkpoint inhibitors (anti-PD-L1 and anti-PD-1) blocking the binding of PD-L1 to PD-1, which allows the T cells to kill tumor cells.")
 compared to the size of a pea (1 cm), a peanut (2 cm), a grape (3 cm), a walnut (4 cm), a lime (5 cm), an egg (6 cm), a peach (7 cm), and a grapefruit (10 cm). Also shown is a 10-cm ruler and a 4-inch ruler.")
 and the dermis (inner layer of the skin). A visible lesion is shown on the skin’s surface. Four numbered blocks show the removal of thin layers of the skin one at a time until all the cancer is removed.")

 is shown passing through the urethra and into the bladder. Fluid is used to fill the bladder. An inset shows a woman lying on an examination table with her knees bent and legs apart. She is covered by a drape. The doctor is looking at an image of the inner wall of the bladder on a computer monitor to check for abnormal areas.")