Pediatric Allogeneic Hematopoietic Stem Cell Transplant (PDQ®)–Health Professional Version
Improved Outcomes After Allogeneic Hematopoietic Stem Cell Transplant (HSCT)
During the past two decades, significant advances have led to improved outcomes after allogeneic HSCT.[1–3] The most significant improvements in survival occurred in unrelated and alternative donor procedures.[4–6] Possible explanations for these improvements in survival include improved patient selection, better supportive care, refined treatment regimens, improved approaches specific to stem cell sources, better intensive care unit experience, and better HLA typing. The sections below focus on modifiable aspects of HSCT, including the optimization of HLA typing and selection of stem cell sources.
References
- Hahn T, McCarthy PL, Hassebroek A, et al.: Significant improvement in survival after allogeneic hematopoietic cell transplantation during a period of significantly increased use, older recipient age, and use of unrelated donors. J Clin Oncol 31 (19): 2437-49, 2013. [PUBMED Abstract]
- Horan JT, Logan BR, Agovi-Johnson MA, et al.: Reducing the risk for transplantation-related mortality after allogeneic hematopoietic cell transplantation: how much progress has been made? J Clin Oncol 29 (7): 805-13, 2011. [PUBMED Abstract]
- Wood WA, Lee SJ, Brazauskas R, et al.: Survival improvements in adolescents and young adults after myeloablative allogeneic transplantation for acute lymphoblastic leukemia. Biol Blood Marrow Transplant 20 (6): 829-36, 2014. [PUBMED Abstract]
- MacMillan ML, Davies SM, Nelson GO, et al.: Twenty years of unrelated donor bone marrow transplantation for pediatric acute leukemia facilitated by the National Marrow Donor Program. Biol Blood Marrow Transplant 14 (9 Suppl): 16-22, 2008. [PUBMED Abstract]
- Harvey J, Green A, Cornish J, et al.: Improved survival in matched unrelated donor transplant for childhood ALL since the introduction of high-resolution matching at HLA class I and II. Bone Marrow Transplant 47 (10): 1294-300, 2012. [PUBMED Abstract]
- Majhail NS, Chitphakdithai P, Logan B, et al.: Significant improvement in survival after unrelated donor hematopoietic cell transplantation in the recent era. Biol Blood Marrow Transplant 21 (1): 142-50, 2015. [PUBMED Abstract]
Allogeneic Hematopoietic Stem Cell Transplant (HSCT) Indications for Hematologic Malignancies
Indications for HSCT vary over time as risk classifications for a given malignancy change and the efficacy of primary therapy improves. It is best to include specific indications in the context of complete therapy for any given disease. With this in mind, links to sections in specific summaries that cover the most common pediatric allogeneic HSCT indications are provided below.
- Acute lymphoblastic leukemia (ALL).
- For more information, see the Treatment of Relapsed Childhood ALL section in Childhood Acute Lymphoblastic Leukemia Treatment.
- Acute myeloid leukemia (AML).
- For more information, see the Treatment of Childhood AML section in Childhood Acute Myeloid Leukemia Treatment.
- Myelodysplastic neoplasms (MDS).
- For more information, see the Treatment of Childhood MDS section in Childhood Myelodysplastic Neoplasms Treatment.
- Juvenile myelomonocytic leukemia (JMML).
- For more information, see the Treatment of JMML section in Juvenile Myelomonocytic Leukemia Treatment.
- Chronic myeloid leukemia (CML).
- For more information, see the Treatment of Recurrent or Refractory Childhood CML section in Childhood Chronic Myeloid Leukemia Treatment.
HLA Matching and Hematopoietic Stem Cell Sources
HLA Overview
Appropriate matching between donor and recipient HLA in the major histocompatibility complex located on chromosome 6 is essential to successful allogeneic hematopoietic stem cell transplant (HSCT) (see Figure 1, Table 1, and Table 2).
 complex; drawing shows the long and short arms of human chromosome 6 with amplification of the HLA region, including the class I A, B, and C alleles, and the class II DP, DQ, and DR alleles.")
HLA class I (A, B, C, etc.) and class II (DRB1, DRB3, DRB4, DRB5, DQB1, DPB1, etc.) alleles are highly polymorphic. Therefore, finding appropriately matched unrelated donors is a challenge for some patients, especially those of certain racial groups (e.g., patients with African, Hispanic, Asian, or Pacific-Islander ancestry).[1,2] Full siblings of cancer patients have a 25% chance of being HLA matched.
Early serological techniques of HLA assessment defined a number of HLA antigens, but more precise DNA methodologies have shown HLA allele-level mismatches in up to 40% of serological HLA antigen matches. These differences are clinically relevant because the use of donors with allele-level mismatches affects survival and rates of graft-versus-host disease (GVHD) to a degree similar to that in patients with antigen-level mismatches.[3] Because of this, DNA-based allele-level HLA typing is standard when unrelated donors are being chosen.
The National Marrow Donor Program has published guidelines for HLA matching. The term for allele-level matching used in their guidelines is antigen recognition domain, which refers to the fact that the allele-level similarities used to define the specific HLA type are associated with areas directly used for antigen recognition. Polymorphisms of the HLA proteins outside of these areas are not involved in the function of these molecules. Therefore, they are often not assessed as part of HLA testing and unlikely to contribute to HLA mismatch.[4]
| Class I Antigens | Class II Antigens | |||||
|---|---|---|---|---|---|---|
| BM = bone marrow; N/A = not applicable; PBSCs = peripheral blood stem cells. | ||||||
| aHLA antigen: A serologically defined, low-resolution method of defining an HLA protein. Differs from allele-level typing at least 40% of the time. Designated by the first two numbers (i.e., for HLA B 35:01, the antigen is HLA B 35). | ||||||
| bHLA allele: A higher-resolution method of defining unique HLA proteins by typing their gene through sequencing or other DNA-based methods that detect unique differences. Designated by at least four numbers (i.e., for HLA B 35:01, 35 is the antigen and 01 is the allele). | ||||||
| cConsensus recommendations for HLA typing, including extended class II typing of mismatched donors, have been published by the National Cancer Institute/National Heart, Lung, and Blood Institute–sponsored Blood and Marrow Transplant Clinical Trials Network.[5] | ||||||
| dSiblings need confirmation that they have fully matched haplotypes with no crossovers in the A to DRB1 region. If parental typing is performed and haplotypes are established, antigen-level typing of class I is adequate. With no parental haplotypes, allele-level typing of eight alleles is recommended. | ||||||
| eParents, cousins, or other family members, with a phenotypic match or near-complete HLA match. | ||||||
| Stem Cell Source | HLA A | HLA B | HLA C | HLA DRB1 | HLA DQB1; HLA DPB1; HLA DRB3,4,5 | |
| Matched siblingd BM/PBSCs | Antigen or allele | Antigen or allele | Optional | Allele | N/A | |
| Mismatched sibling/other related-donore BM/PBSCs | Allele | Allele | Allele | Allele | Recommended | |
| Unrelated-donor BM/PBSCs | Allele | Allele | Allele | Allele | Recommended | |
| Unrelated-donor cord blood | Antigen (allele recommended) | Antigen (allele recommended) | Allele recommended | Allele | N/A | |
| If These HLA Antigens and Alleles Match: | Then the Donor Is Considered to be This Type of Match: |
|---|---|
| A, B, and DRB1 | 6/6 |
| A, B, C, and DRB1 | 8/8 |
| A, B, C, DRB1, and DQB1 | 10/10 |
| A, B, C, DRB1, DQB1, and DPB1 | 12/12 |
HLA Matching Considerations for Sibling and Related Donors
The most commonly used related donor is a sibling from the same parents who, at a minimum, is HLA matched for HLA A, HLA B, and HLA DRB1 at the antigen level. Given the distance between HLA A and HLA DRB1 on chromosome 6, there is approximately a 1% possibility of a crossover event occurring in a possible sibling match. Because a crossover event could involve the HLA C antigen and because parents may share HLA antigens that actually differ at the allele level, many centers perform allele-level typing of possible sibling donors at all of the key HLA antigens (HLA A, B, C, and DRB1). Any related donor that is not a full sibling should have full HLA typing because similar haplotypes from different parents could differ at the allele level.
Although single-antigen mismatched related donors (5/6 antigen matched) were used interchangeably with matched sibling donors in some studies, a large Center for International Blood and Marrow Transplant Research (CIBMTR) study in pediatric HSCT recipients showed that the use of 5/6 antigen-matched related donors resulted in rates of GVHD and overall survival (OS) equivalent to rates in 8/8-allele-level-matched unrelated donors and slightly inferior survival than in fully matched siblings.[6] Any siblings with single mismatches should have extended typing to ensure that if the mismatch is caused by a crossover, it only occurs with one antigen. If clinicians choose siblings with multiple antigen mismatches as donors, haploidentical approaches may be warranted.
HLA Matching Considerations for Unrelated Donors
Optimal outcomes are achieved in unrelated allogeneic bone marrow transplant when the pairs of antigens at HLA A, B, C, and DRB1 are matched between the donor and the recipient at the allele level (termed an 8/8 match) (see Table 2).[7] A single antigen/allele mismatch at any of these antigens (7/8 match) lowers the probability of survival between 5% and 10%, with a similar increase in the amount of significant (grades III–IV) acute GVHD.[7] Of these four antigen pairs, different reports have shown HLA A, C, and DRB1 mismatches to potentially be more highly associated with mortality than the other antigens,[3,7,8] but the differences in outcome are small and inconsistent, making it very difficult to conclude that one can pick a more favorable mismatch by choosing one type of antigen mismatch over another. Many study groups are attempting to define specific antigens or pairs of antigens that are associated with either good or poor outcomes. For example, a specific HLA C mismatch (HLA-C*03:03/03:04) produces outcomes similar to a match. Therefore, selection of this mismatch is desirable in an otherwise matched donor/pair combination.[9]
It is well understood that class II antigen DRB1 mismatches increase GVHD incidence and worsen survival.[8] Subsequent data have also shown that multiple mismatches of DQB1, DPB1, and DRB3,4,5 lead to worse outcomes in the setting of less-than-8/8 matches.[10] DPB1 mismatches have been extensively studied and classified as permissive or nonpermissive on the basis of T-cell epitope matching. Patients with 10/10 matches and nonpermissive DPB1 mismatches have more transplant-related mortality but have survival rates similar to those with DPB1 matches or permissive matches. Those with 9/10 matches who have nonpermissive DPB1 mismatches have worse survival than those with permissive mismatches or DPB1 matches.[11–13]
With these findings in mind, a 7/8- or 8/8-matched unrelated donor can be used routinely. However, outcomes may be further improved with the following:
- Extended typing of DQB1, DPB1, and DRB3,4,5.[4,11–13]
- Extended HLA testing to select appropriate donors in the context of HLA-sensitized patients to avoid the potential risk of graft failure.[14,15] HLA sensitization is detected by testing for the presence of specific anti-HLA antibodies and avoiding donors who have any HLA antigens associated with the antibodies present in the recipient.
- Use of younger donors.[5]
- Matching cytomegalovirus (CMV)-positive recipients with CMV-positive donors and matching CMV-negative recipients with CMV-negative donors.[16]
- Use of blood type–compatible unrelated donors.[5]
; a mismatch (0201 for both donor and recipient and 0201 for donor, 0401 for recipient) shown by an arrow pointing in a direction that promotes GVHD (GVH-O); a mismatch (0201 for both donor and recipient and 0401 for donor, 0201 for recipient) shown by an arrow pointing in a direction that promotes rejection (R-O); and a bidirectional mismatch (0201 for donor, 0301 for recipient, and 0401 for both donor and recipient) shown by arrows pointing in two directions, a direction that promotes rejection (R-O) and a direction that promotes GVHD (GVH-O).")
If a donor or recipient has a duplication of one of their HLA alleles, they will have a half match and a mismatch only in one direction. Figure 2 illustrates that these mismatches will occur in either a direction that promotes GVHD (GVH-O, donor cells can detect a mismatch in a recipient which could cause GVHD) or a direction that promotes rejection (R-O, recipient cells can detect a mismatch in a donor that could lead to rejection). When 8/8-matched unrelated donors are compared with 7/8 donors mismatched in the GVH-O direction, 7/8 mismatched in the R-O direction, or 7/8 mismatched in both directions, the mismatch in the R-O direction leads to rates of grades III and IV acute GVHD similar to rates in the 8/8 matched and better than in the other two combinations. The 7/8 mismatched in only the R-O direction is preferred over GVH-O and bidirectional mismatches.[17] It is important to note that this observation in unrelated donors differs from observations in cord blood recipients, outlined below.
HLA Matching and Cell Dose Considerations for Unrelated Cord Blood HSCT
Another commonly used hematopoietic stem cell source is unrelated umbilical cord blood, which is harvested from donor placentas moments after birth. The cord blood is processed, HLA typed, cryopreserved, and banked.
Unrelated cord blood transplant has been successful with less-stringent HLA matching requirements compared with standard related or unrelated donors, probably because of limited antigen exposure experienced in utero and different immunological composition. Cord blood matching has traditionally been performed at an intermediate level for HLA A and B and at an allele level (high resolution) for DRB1. However, as outlined below, more extended typing can be helpful.
Although better outcomes occur when 6/6 or 5/6 HLA-matched units are used,[18] successful HSCT has occurred even with 4/6 or less HLA-matched units. In a large CIBMTR/Eurocord study, better matching at the allele level using eight antigens (matching for HLA A, B, C, and DRB1) resulted in less transplant-related mortality and improved survival. Best outcome was noted with 8/8 allele matching versus 4/8 to 7/8 matches, with poor survival in patients with five or more allele mismatches. Patients receiving 8/8-matched cord blood did not require higher cell doses for better outcomes. However, patients with one to three allele mismatches had less transplant-related mortality with total nucleated cell counts higher than 3 × 107/kg, and those with four allele mismatches required a total nucleated cell count higher than 5 × 107/kg to decrease transplant-related mortality.[19] This observation was noted to be especially important in cord blood transplants for nonmalignant disorders, where any mismatching below 7/8 alleles led to inferior survival.[20] Many centers will type additional alleles and use the best match possible, but the impact of DQB1, DPB1, and DRB3,4,5 mismatches has not been studied in detail.
As in unrelated peripheral blood stem cells (PBSCs) or bone marrow donors, extended HLA testing can support the selection of appropriate cord blood units in HLA-sensitized patients to avoid the potential risk of graft failure.[21,22] Evidence also suggests that selecting a mismatched cord blood unit, where the mismatch involves a noninherited maternal antigen, may improve survival.[23,24]
As with unrelated donors, individuals can occasionally have duplicate HLA alleles (e.g., the HLA A allele is 01:01 on both chromosomes). When this occurs in a donor product and the allele is matched to one of the recipient alleles, the recipient immune response will see the donor allele as matched (matched, in the rejection direction), but the donor immune response will see a mismatch in the recipient (mismatched in the GVHD direction). This variation of partial mismatching has been shown to be important in cord blood transplant outcomes. Mismatches that are only in the GVHD direction (i.e., GVH-O) lead to lower transplant-related mortality and overall mortality than those with rejection direction only (i.e., R-O) mismatches.[25] R-O mismatches have outcomes similar to those of bidirectional mismatches.[26] Although these studies suggest that using unidirectional mismatching as a criteria for cord blood selection may be beneficial, a Eurocord–European Society for Blood and Marrow Transplantation analysis disputes the value of this type of mismatching.[27]
Two aspects of umbilical cord blood HSCT have made the practice more widely applicable. First, because a successful procedure can occur with multiple HLA mismatches, more than 95% of patients from a wide variety of ethnicities are able to find at least a 4/6-matched cord blood unit.[1,28] Second, as mentioned above, adequate cell dose (minimum 2.5–3 × 107 total nucleated cells/kg and 1.5 × 105 CD34+ cells/kg) has been shown to be associated with improved survival.[29,30] Total nucleated cells are generally used to judge units because techniques to measure CD34-positive doses have not been standardized. Because even large single umbilical cord blood units are only able to supply these minimum doses to recipients weighing up to 40 kg to 50 kg, early umbilical cord blood HSCT focused mainly on smaller children. Later studies showed that barriers of this smaller size could be overcome by using two umbilical cord blood units if each of the units is at least a 4/6 HLA match with the recipient. Because two cord blood units provide higher cell doses, umbilical cord blood transplant is now used widely for larger children and adults.[31]
If a single unit provides an adequate cell dose, there may be disadvantages to adding a second unit.[32][Level of evidence A1] Two randomized trials showed that in children who had adequately sized single units, the addition of a second unit did not alter relapse, transplant-related mortality, or survival rates, but was associated with higher rates of extensive chronic GVHD.[32,33]
Investigators have shown that by using combinations of cytokines and other compounds to expand cord blood before infusion, engraftment of cord blood cells can occur more rapidly than after standard approaches.[34–37] Although some studies that used multiple units or split units showed that expanded units will engraft early and then give way to nonexpanded units for long-term reconstitution,[38] other studies are showing persistence of expanded cells, implying preservation of stem cells through the expansion process.[36,37] A number of these approaches are under investigation. The U.S. Food and Drug Administration (FDA) approved an approach that uses a single unit split into two fractions, expanding one in culture with nicotinamide and infusing the second fraction without manipulation (omidubicel). One randomized trial compared omidubicel with standard cord blood transplant. Patients who received omidubicel had faster neutrophil and platelet engraftment, fewer bacterial and fungal infections, and fewer hospital days in the first 3 months after HSCT.[39] Notably, there was no difference in survival and GVHD outcomes.
Haploidentical HSCT
Early HSCT studies demonstrated progressively higher percentages of patients experiencing severe GVHD and lower survival rates as the number of donor/recipient HLA mismatches increased.[40] Other studies showed that even with very high numbers of donors in unrelated-donor registries, patients with rare HLA haplotypes and patients with certain ethnic backgrounds (e.g., patients with African, Hispanic, Asian, or Pacific-Islander ancestry) have a low chance of achieving desired levels of HLA matching (7/8 or 8/8 match at the allele level).[2]
To allow access to HSCT for patients without fully HLA-matched donor options, investigators have developed techniques allowing the use of siblings, parents, or other relatives who share only a single haplotype of the HLA complex with the patient and are thus half matches. Most approaches developed to date rely on T-cell depletion of the product before infusion into the patient. The main challenge associated with this approach is intense immune suppression with delayed immune recovery, which can result in lethal infections,[41] increased risk of Epstein-Barr virus (EBV)–associated lymphoproliferative disorder, and high rates of relapse.[42] This led to inferior survival compared with matched-donor HSCTs in the past and resulted in the procedure being used mainly at larger academic centers with a specific research focus on studying and developing this approach.
Improvements in haploidentical approaches, however, have resulted in better outcomes, with many groups reporting survival similar to that of unrelated marrow or cord blood approaches.[43–46] These improvements include the following:
- Newer techniques of T-cell depletion and add-back of specific cell populations (e.g., CD3 or alpha-beta CD3/CD19-negative selection) have decreased transplant-related mortality.[47]; [45,46,48,49][Level of evidence C2]
- Reduced toxicity preparative regimens have led to decreased transplant-related mortality.[45,50]
- Better supportive care has decreased the chance of morbidity from infection or EBV-associated lymphoproliferative disorder.[51]
- Patient-donor combinations that have specific killer immunoglobulin-like receptor mismatches have shown decreased likelihood of relapse in some studies. For more information, see the Role of killer immunoglobulin-like receptor (KIR) mismatching in HSCT section.
- Certain techniques, such as using combinations of granulocyte colony-stimulating factor–primed bone marrow and PBSCs with posttransplant antibody–based T-cell depletion [52] or post-HSCT cyclophosphamide (chemotherapeutic T-cell depletion),[45,46,53]; [54][Level of evidence C1] have made these procedures more accessible because they do not use the expensive and complicated processing necessary for traditional T-cell depletion.
Reported survival rates using many different types of haploidentical approaches range from 25% to 80%, depending on the technique and the risk of the patient undergoing the procedure.[42,43,52,53]; [54][Level of evidence C1] Retrospective trials in adults have shown similar outcomes after haploidentical-donor transplants compared with matched-unrelated donor or cord blood transplants.[55,56] One prospective randomized trial in adults with hematologic malignancies that used reduced-intensity regimens showed similar progression-free survival, but lower relapse rates and better OS using haploidentical donors.[57] Pediatric trials using haploidentical donors have shown better outcomes with myeloablative preparative regimens, and survival is comparable to nonhaploidentical approaches.[45,46,49,58] One prospective trial in pediatric patients showed that disease-free survival (DFS) was superior using haploidentical approaches compared with mismatched unrelated-donor approaches. DFS rates in patients treated with haploidentical approaches were similar to those in patients treated with other stem cell sources.[45]
Patients who have been sensitized to HLA antigens and develop antibodies to HLA alleles that are present among the mismatched alleles of their haploidentical donor are at increased risk of rejection of their haploidentical graft. Clinicians should choose donors with HLA types against whom the recipient does not have an antibody present, if possible. Guidelines on how to best approach this issue have been published.[59]
Comparison of Stem Cell Products
Currently, the following three stem cell products are used from both related and unrelated donors:
- Bone marrow.
- PBSCs.
- Cord blood.
Bone marrow or PBSCs, including partially HLA-matched (half or more antigens [haploidentical]) related bone marrow or PBSCs, can be used after in vitro or in vivo T-cell depletion, and these products behave differently from other stem cell products. A comparison of stem cell products is presented in Table 3.
| PBSCs | BM | Cord Blood | T-cell–Depleted BM/PBSCs | Haploidentical T-cell–Depleted BM/PBSCs | |
|---|---|---|---|---|---|
| BM = bone marrow; EBV-LPD = Epstein-Barr virus–associated lymphoproliferative disorder; GVHD = graft-versus-host disease; HSCT = hematopoietic stem cell transplant; PBSCs = peripheral blood stem cells. | |||||
| aAssuming no development of GVHD. If patients develop GVHD, immune reconstitution is delayed until resolution of the GVHD and discontinuation of immune suppression. | |||||
| bIf a haploidentical donor is used, longer times to immune reconstitution may occur. | |||||
| T-cell content | High | Moderate | Low | Very low | Very low |
| CD34+ content | Moderate–high | Moderate | Low (but higher potency) | Moderate–high | Moderate–high |
| Time to neutrophil recovery | Rapid: median, 16 d (11–29 d) [60] | Moderate: median, 21 d (12–35 d) [60] | Slower: median, 23 d (11–133 d) [33] | Rapid: median, 16 d (9–40 d) [61] | Rapid: median, 13 d (10–20 d) [62] |
| Early post-HSCT risk of infections, EBV-LPD | Low–moderate | Moderate | High | Very high | Very high |
| Risk of graft rejection | Low | Low–moderate | Moderate–high | Moderate–high | Moderate–high |
| Time to immune reconstitutiona | Rapid (6–12 mo) | Moderate (6–18 mo) | Slow (6–24 mo) | Slow (6–24 mo) | Slow (9–24 mo)b |
| Risk of acute GVHD | Moderate | Moderate | Moderate | Low | Low |
| Risk of chronic GVHD | High | Moderate | Low | Low | Low |
The main differences between the products are the numbers of T cells and CD34-positive progenitor cells present. Very high levels of T cells are present in PBSCs, intermediate numbers in bone marrow, and low numbers in cord blood and T-cell–depleted products. Patients receiving T-cell–depleted products or cord blood generally have slower hematopoietic recovery, increased risk of infection, late immune reconstitution, higher risks of nonengraftment, and increased risk of EBV-associated lymphoproliferative disorder. This is countered by lower rates of GVHD and an ability to offer transplant to patients for whom full HLA matching is not available. Higher doses of T cells and other cells in PBSCs result in rapid neutrophil recovery and immune reconstitution but also increase rates of chronic GVHD.
Only a few studies have directly compared outcomes of different stem cell sources/products in pediatric patients.
Evidence (comparison of outcomes of stem cell sources/products in children):
- A retrospective registry study of pediatric patients who underwent HSCT for acute leukemia compared those who received related-donor bone marrow with those who received related-donor PBSCs.[63]
- Although the bone marrow and PBSC recipient cohorts differed some in their risk profiles, after statistical correction, increased risk of GVHD and transplant-related mortality associated with PBSCs led to poorer survival in the PBSC group.
- A retrospective study of Japanese children with acute leukemia compared 90 children who received PBSCs with 571 children who received bone marrow.[64]
- The study confirmed higher transplant-related mortality caused by GVHD and inferior survival among the children who received PBSCs.
- A large Blood and Marrow Transplant Clinical Trials Network trial for patients requiring unrelated donors included a number of pediatric patients. Patients were randomly assigned to receive either bone marrow or PBSCs. This trial demonstrated the following:[65]
- OS was identical using either source, but rates of chronic GVHD were significantly higher in the PBSC arm, with a small increase in rejection in the bone marrow arm.
- Rejections were rare in pediatric patients.
- There was an insufficient number of patients to draw specific conclusions about rejection risk in children who received bone marrow.
These reports, combined with a lack of prospective studies comparing bone marrow and PBSCs, have led most pediatric transplant protocols to prefer bone marrow over PBSCs from related donors. This approach is further supported by a meta-analysis that included additional retrospective trials.[66]
Published studies comparing unrelated cord blood and bone marrow have been retrospective, with weaknesses inherent in such analyses.
Evidence (comparison of unrelated cord blood versus bone marrow outcomes):
- In one study, pediatric patients with acute lymphoblastic leukemia (ALL) who underwent HSCT and received 8/8 HLA-matched unrelated-donor bone marrow were compared with those who received unrelated cord blood.[18]
- The analysis showed that the best survival occurred in recipients of 6/6 HLA-matched cord blood.
- Survival after 8/8 HLA-matched unrelated bone marrow was slightly worse and was statistically identical to survival for patients receiving 5/6 and 4/6 HLA-matched cord blood units.
- In another study from a single center consisting of mostly adult patients with acute myeloid leukemia (AML), myelodysplastic neoplasms (MDS), and ALL, outcomes for cord blood recipients were compared with outcomes for recipients of matched and mismatched unrelated-donor bone marrow/PBSCs.[67]
- Better survival because of less relapse was noted in cord blood recipients. This result was mainly due to superior survival in patients with minimal residual disease (MRD) present just before transplant.
- No difference was seen in relapse or survival between patients with pre-HSCT MRD and patients without pre-HSCT MRD if they received cord blood.
- These results are controversial because they contradict many other studies that showed that the presence of pre-HSCT MRD in cord blood recipients led to increased relapse and inferior survival rates.[68–71]
- The CIBMTR compared outcomes of children with low-risk and intermediate-risk ALL and AML who underwent transplant between 2000 and 2014 using alternative donors (non–HLA-matched related or unrelated), including 7/8 HLA-matched bone marrow (n = 172) and 4/6 or greater HLA-matched umbilical cord blood (n = 1,613).[72]
- In multivariate analysis, patients who received 7/8 HLA-matched bone marrow versus umbilical cord blood had similar GVHD-free, relapse-free survival (hazard ratio [HR], 1.12; 95% confidence interval [CI], 0.87–1.45; P = .39), chronic GVHD-free, relapse-free survival (HR, 1.06; 95% CI, 0.82–1.38; P = .66), and OS (HR, 1.07; 95% CI, 0.80–1.44; P = .66).
- Relapse may have been higher in the 7/8 HLA-matched bone marrow group (HR, 1.44; 95% CI, 1.03–2.02; P = .03; the publication called this a trend as they chose a cutoff value of 0.01% to control for multiple comparisons).
- The patients in the 7/8 HLA-matched bone marrow group had a significantly higher risk of grades III to IV acute GVHD (HR, 1.70; 95% CI, 1.16–2.48; P = .006) and chronic GVHD (HR, 6.17; 95% CI, 2.2–17.33; P = .0006) than did the patients in the umbilical cord blood group.
On the basis of these studies, most transplant centers consider matched sibling bone marrow to be the preferred stem cell source/product. If a sibling donor is not available, fully matched unrelated-donor bone marrow, HLA-matched (4/6 to 6/6 or 6/8 to 8/8) cord blood from a single unit with an adequate cell dose, or a haploidentical HSCT lead to similar survival rates.[49][Level of evidence C2] For more information about the prevention of acute GVHD, see the Prevention and treatment of acute GVHD section in Complications, Graft-Versus-Host Disease, and Late Effects After Pediatric Hematopoietic Stem Cell Transplant.
Other Donor Characteristics Associated With Outcome
HLA matching has consistently been the most important factor associated with improved survival in allogeneic HSCT, but a number of other donor characteristics have been shown to affect key outcomes. Higher cell dose from the donor has also been shown to be important when related, unrelated, or haploidentical bone marrow or PBSC donors are used.[73,74] For more information, see the HLA Matching and Cell Dose Considerations for Unrelated Cord Blood HSCT section. The effects of donor age, blood type, CMV status, sex, and parity of female donors have also been studied.
Ideally, after HLA matching, transplant centers should select donors based on the following characteristics:
- Donor age. The youngest donor available is generally preferred (when considering pediatric donors, the size and ability to obtain an adequate cell dose comes into consideration).[75,76]
- CMV status of the recipient. CMV-negative donor matched to CMV-negative recipient and CMV-positive donor matched to CMV-positive recipient are preferred.[77]
- Donor blood type. Matching of blood type between donor and recipient is preferred, although not required. If only blood type–mismatched donors are available, a minor mismatch is preferred over a major mismatch.[78–80]
- Donor sex and parity of female donors. Male or nonparous female donors are preferred over parous female donors.[76,81]
It is rare for a donor-recipient pair to fit perfectly into this algorithm, and determining which of these characteristics should be chosen over others has been controversial.
Evidence (donor-recipient characteristics):
- A CIBMTR study examined 6,349 patients who underwent transplant for hematological malignancies from 1988 to 2006. The study tested the effect of donor characteristics while adjusting for disease risk and other key transplant characteristics. The data from this study showed the following:[76]
- In addition to HLA mismatching, older donor age and major or minor ABO blood type mismatching increased overall mortality.
- Parous female graft recipients experienced lower rates of relapse.
- Recipients of younger donor grafts had lower rates of acute GVHD.
- Recipients of parous female grafts had higher rates of chronic GVHD.
- Recipient CMV status was more important than donor CMV status (recipients who are CMV positive are at higher risk of mortality independent of the donor CMV status), although a CMV-negative donor to a CMV-negative recipient combination improves survival.
- A cohort of 4,690 patients who underwent transplant between 2007 and 2011 was tested by a multivariate analysis for independent predictors of survival in an EBMT study. The study demonstrated the following:[82]
- Older donor age was confirmed to be independently associated with worse OS; every 10 years of donor age increased the risk of mortality by 5.5%.
- HLA matching continued to have the most important effect on survival; ABO mismatching was not confirmed to have a continuing effect.
- A study of over 10,000 matched donor-recipient pairs attempted to define a hierarchy that could prioritize the non-HLA characteristics (donor age, sex, blood type, CMV status, etc.) that have been described to affect outcomes.[83]
- Although the study was unable to create a hierarchical algorithm of modifiable factors, it showed that, by far, younger donor age is the most important factor. The study found a decrease in OS of 3% for every 10-year increment of increased donor age.
Thus, after HLA matching, donor age is likely the most important factor to optimize. Of note, if the recipient is CMV negative, finding a CMV-negative donor is also a high priority.
Several studies have attempted to identify characteristics of the best donors for haploidentical procedures. As with conventional bone marrow transplant, use of younger donors appears to be beneficial, but data regarding donor sex are inconclusive. Studies involving intense T-cell depletion have noted better outcomes using maternal donors,[84] but studies using posttransplant cyclophosphamide or intense immune suppression seem to favor male donors.[85,86] Further study is needed to clarify this important issue.
One large comparison of haploidentical donors showed an effect of ABO incompatibility on engraftment (risk of rejection doubling from 6% to 12%, ABO match vs. ABO major mismatch), and patients receiving bidirectionally mismatched donors had a 2.4-fold increase in grades II to IV acute GVHD.[87] As with nonhaploidentical donors, significant improvement of outcomes was noted when younger donors were used for haploidentical procedures compared with older donors, with an HR of 1.13 for each decade of life that the donor is older.[88]
References
- Barker JN, Byam CE, Kernan NA, et al.: Availability of cord blood extends allogeneic hematopoietic stem cell transplant access to racial and ethnic minorities. Biol Blood Marrow Transplant 16 (11): 1541-8, 2010. [PUBMED Abstract]
- Gragert L, Eapen M, Williams E, et al.: HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med 371 (4): 339-48, 2014. [PUBMED Abstract]
- Woolfrey A, Klein JP, Haagenson M, et al.: HLA-C antigen mismatch is associated with worse outcome in unrelated donor peripheral blood stem cell transplantation. Biol Blood Marrow Transplant 17 (6): 885-92, 2011. [PUBMED Abstract]
- Dehn J, Spellman S, Hurley CK, et al.: Selection of unrelated donors and cord blood units for hematopoietic cell transplantation: guidelines from the NMDP/CIBMTR. Blood 134 (12): 924-934, 2019. [PUBMED Abstract]
- Howard CA, Fernandez-Vina MA, Appelbaum FR, et al.: Recommendations for donor human leukocyte antigen assessment and matching for allogeneic stem cell transplantation: consensus opinion of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biol Blood Marrow Transplant 21 (1): 4-7, 2015. [PUBMED Abstract]
- Shaw PJ, Kan F, Woo Ahn K, et al.: Outcomes of pediatric bone marrow transplantation for leukemia and myelodysplasia using matched sibling, mismatched related, or matched unrelated donors. Blood 116 (19): 4007-15, 2010. [PUBMED Abstract]
- Flomenberg N, Baxter-Lowe LA, Confer D, et al.: Impact of HLA class I and class II high-resolution matching on outcomes of unrelated donor bone marrow transplantation: HLA-C mismatching is associated with a strong adverse effect on transplantation outcome. Blood 104 (7): 1923-30, 2004. [PUBMED Abstract]
- Petersdorf EW, Kollman C, Hurley CK, et al.: Effect of HLA class II gene disparity on clinical outcome in unrelated donor hematopoietic cell transplantation for chronic myeloid leukemia: the US National Marrow Donor Program Experience. Blood 98 (10): 2922-9, 2001. [PUBMED Abstract]
- Fernandez-Viña MA, Wang T, Lee SJ, et al.: Identification of a permissible HLA mismatch in hematopoietic stem cell transplantation. Blood 123 (8): 1270-8, 2014. [PUBMED Abstract]
- Fernández-Viña MA, Klein JP, Haagenson M, et al.: Multiple mismatches at the low expression HLA loci DP, DQ, and DRB3/4/5 associate with adverse outcomes in hematopoietic stem cell transplantation. Blood 121 (22): 4603-10, 2013. [PUBMED Abstract]
- Fleischhauer K, Shaw BE, Gooley T, et al.: Effect of T-cell-epitope matching at HLA-DPB1 in recipients of unrelated-donor haemopoietic-cell transplantation: a retrospective study. Lancet Oncol 13 (4): 366-74, 2012. [PUBMED Abstract]
- Crocchiolo R, Zino E, Vago L, et al.: Nonpermissive HLA-DPB1 disparity is a significant independent risk factor for mortality after unrelated hematopoietic stem cell transplantation. Blood 114 (7): 1437-44, 2009. [PUBMED Abstract]
- Pidala J, Lee SJ, Ahn KW, et al.: Nonpermissive HLA-DPB1 mismatch increases mortality after myeloablative unrelated allogeneic hematopoietic cell transplantation. Blood 124 (16): 2596-606, 2014. [PUBMED Abstract]
- Spellman S, Bray R, Rosen-Bronson S, et al.: The detection of donor-directed, HLA-specific alloantibodies in recipients of unrelated hematopoietic cell transplantation is predictive of graft failure. Blood 115 (13): 2704-8, 2010. [PUBMED Abstract]
- Ciurea SO, Thall PF, Wang X, et al.: Donor-specific anti-HLA Abs and graft failure in matched unrelated donor hematopoietic stem cell transplantation. Blood 118 (22): 5957-64, 2011. [PUBMED Abstract]
- Shaw BE, Mayor NP, Szydlo RM, et al.: Recipient/donor HLA and CMV matching in recipients of T-cell-depleted unrelated donor haematopoietic cell transplants. Bone Marrow Transplant 52 (5): 717-725, 2017. [PUBMED Abstract]
- Hurley CK, Woolfrey A, Wang T, et al.: The impact of HLA unidirectional mismatches on the outcome of myeloablative hematopoietic stem cell transplantation with unrelated donors. Blood 121 (23): 4800-6, 2013. [PUBMED Abstract]
- Eapen M, Rubinstein P, Zhang MJ, et al.: Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet 369 (9577): 1947-54, 2007. [PUBMED Abstract]
- Eapen M, Klein JP, Ruggeri A, et al.: Impact of allele-level HLA matching on outcomes after myeloablative single unit umbilical cord blood transplantation for hematologic malignancy. Blood 123 (1): 133-40, 2014. [PUBMED Abstract]
- Eapen M, Wang T, Veys PA, et al.: Allele-level HLA matching for umbilical cord blood transplantation for non-malignant diseases in children: a retrospective analysis. Lancet Haematol 4 (7): e325-e333, 2017. [PUBMED Abstract]
- Takanashi M, Atsuta Y, Fujiwara K, et al.: The impact of anti-HLA antibodies on unrelated cord blood transplantations. Blood 116 (15): 2839-46, 2010. [PUBMED Abstract]
- Cutler C, Kim HT, Sun L, et al.: Donor-specific anti-HLA antibodies predict outcome in double umbilical cord blood transplantation. Blood 118 (25): 6691-7, 2011. [PUBMED Abstract]
- Rocha V, Spellman S, Zhang MJ, et al.: Effect of HLA-matching recipients to donor noninherited maternal antigens on outcomes after mismatched umbilical cord blood transplantation for hematologic malignancy. Biol Blood Marrow Transplant 18 (12): 1890-6, 2012. [PUBMED Abstract]
- van Rood JJ, Stevens CE, Smits J, et al.: Reexposure of cord blood to noninherited maternal HLA antigens improves transplant outcome in hematological malignancies. Proc Natl Acad Sci U S A 106 (47): 19952-7, 2009. [PUBMED Abstract]
- Kanda J, Atsuta Y, Wake A, et al.: Impact of the direction of HLA mismatch on transplantation outcomes in single unrelated cord blood transplantation. Biol Blood Marrow Transplant 19 (2): 247-54, 2013. [PUBMED Abstract]
- Stevens CE, Carrier C, Carpenter C, et al.: HLA mismatch direction in cord blood transplantation: impact on outcome and implications for cord blood unit selection. Blood 118 (14): 3969-78, 2011. [PUBMED Abstract]
- Cunha R, Loiseau P, Ruggeri A, et al.: Impact of HLA mismatch direction on outcomes after umbilical cord blood transplantation for hematological malignant disorders: a retrospective Eurocord-EBMT analysis. Bone Marrow Transplant 49 (1): 24-9, 2014. [PUBMED Abstract]
- Barker JN, Rocha V, Scaradavou A: Optimizing unrelated donor cord blood transplantation. Biol Blood Marrow Transplant 15 (1 Suppl): 154-61, 2009. [PUBMED Abstract]
- Wagner JE, Barker JN, DeFor TE, et al.: Transplantation of unrelated donor umbilical cord blood in 102 patients with malignant and nonmalignant diseases: influence of CD34 cell dose and HLA disparity on treatment-related mortality and survival. Blood 100 (5): 1611-8, 2002. [PUBMED Abstract]
- Rubinstein P, Carrier C, Scaradavou A, et al.: Outcomes among 562 recipients of placental-blood transplants from unrelated donors. N Engl J Med 339 (22): 1565-77, 1998. [PUBMED Abstract]
- Barker JN, Weisdorf DJ, DeFor TE, et al.: Transplantation of 2 partially HLA-matched umbilical cord blood units to enhance engraftment in adults with hematologic malignancy. Blood 105 (3): 1343-7, 2005. [PUBMED Abstract]
- Michel G, Galambrun C, Sirvent A, et al.: Single- vs double-unit cord blood transplantation for children and young adults with acute leukemia or myelodysplastic syndrome. Blood 127 (26): 3450-7, 2016. [PUBMED Abstract]
- Wagner JE, Eapen M, Carter S, et al.: One-unit versus two-unit cord-blood transplantation for hematologic cancers. N Engl J Med 371 (18): 1685-94, 2014. [PUBMED Abstract]
- Stiff PJ, Montesinos P, Peled T, et al.: Cohort-Controlled Comparison of Umbilical Cord Blood Transplantation Using Carlecortemcel-L, a Single Progenitor-Enriched Cord Blood, to Double Cord Blood Unit Transplantation. Biol Blood Marrow Transplant 24 (7): 1463-1470, 2018. [PUBMED Abstract]
- Anand S, Thomas S, Hyslop T, et al.: Transplantation of Ex Vivo Expanded Umbilical Cord Blood (NiCord) Decreases Early Infection and Hospitalization. Biol Blood Marrow Transplant 23 (7): 1151-1157, 2017. [PUBMED Abstract]
- Wagner JE, Brunstein CG, Boitano AE, et al.: Phase I/II Trial of StemRegenin-1 Expanded Umbilical Cord Blood Hematopoietic Stem Cells Supports Testing as a Stand-Alone Graft. Cell Stem Cell 18 (1): 144-55, 2016. [PUBMED Abstract]
- Horwitz ME, Wease S, Blackwell B, et al.: Phase I/II Study of Stem-Cell Transplantation Using a Single Cord Blood Unit Expanded Ex Vivo With Nicotinamide. J Clin Oncol 37 (5): 367-374, 2019. [PUBMED Abstract]
- Delaney C, Heimfeld S, Brashem-Stein C, et al.: Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med 16 (2): 232-6, 2010. [PUBMED Abstract]
- Horwitz ME, Stiff PJ, Cutler C, et al.: Omidubicel vs standard myeloablative umbilical cord blood transplantation: results of a phase 3 randomized study. Blood 138 (16): 1429-1440, 2021. [PUBMED Abstract]
- Beatty PG, Clift RA, Mickelson EM, et al.: Marrow transplantation from related donors other than HLA-identical siblings. N Engl J Med 313 (13): 765-71, 1985. [PUBMED Abstract]
- Aversa F, Tabilio A, Velardi A, et al.: Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med 339 (17): 1186-93, 1998. [PUBMED Abstract]
- Barrett J, Gluckman E, Handgretinger R, et al.: Point-counterpoint: haploidentical family donors versus cord blood transplantation. Biol Blood Marrow Transplant 17 (1 Suppl): S89-93, 2011. [PUBMED Abstract]
- Leung W, Campana D, Yang J, et al.: High success rate of hematopoietic cell transplantation regardless of donor source in children with very high-risk leukemia. Blood 118 (2): 223-30, 2011. [PUBMED Abstract]
- González-Vicent M, Molina B, Andión M, et al.: Allogeneic hematopoietic transplantation using haploidentical donor vs. unrelated cord blood donor in pediatric patients: a single-center retrospective study. Eur J Haematol 87 (1): 46-53, 2011. [PUBMED Abstract]
- Pulsipher MA, Ahn KW, Bunin NJ, et al.: KIR-favorable TCR-αβ/CD19-depleted haploidentical HCT in children with ALL/AML/MDS: primary analysis of the PTCTC ONC1401 trial. Blood 140 (24): 2556-2572, 2022. [PUBMED Abstract]
- Merli P, Algeri M, Galaverna F, et al.: TCRαβ/CD19 cell-depleted HLA-haploidentical transplantation to treat pediatric acute leukemia: updated final analysis. Blood 143 (3): 279-289, 2024. [PUBMED Abstract]
- Handgretinger R, Chen X, Pfeiffer M, et al.: Feasibility and outcome of reduced-intensity conditioning in haploidentical transplantation. Ann N Y Acad Sci 1106: 279-89, 2007. [PUBMED Abstract]
- Locatelli F, Merli P, Pagliara D, et al.: Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130 (5): 677-685, 2017. [PUBMED Abstract]
- Bertaina A, Zecca M, Buldini B, et al.: Unrelated donor vs HLA-haploidentical α/β T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood 132 (24): 2594-2607, 2018. [PUBMED Abstract]
- Bethge WA, Faul C, Bornhäuser M, et al.: Haploidentical allogeneic hematopoietic cell transplantation in adults using CD3/CD19 depletion and reduced intensity conditioning: an update. Blood Cells Mol Dis 40 (1): 13-9, 2008. [PUBMED Abstract]
- Leen AM, Christin A, Myers GD, et al.: Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood 114 (19): 4283-92, 2009. [PUBMED Abstract]
- Huang XJ, Liu DH, Liu KY, et al.: Haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion for the treatment of hematological malignancies. Bone Marrow Transplant 38 (4): 291-7, 2006. [PUBMED Abstract]
- Luznik L, Fuchs EJ: High-dose, post-transplantation cyclophosphamide to promote graft-host tolerance after allogeneic hematopoietic stem cell transplantation. Immunol Res 47 (1-3): 65-77, 2010. [PUBMED Abstract]
- Berger M, Lanino E, Cesaro S, et al.: Feasibility and Outcome of Haploidentical Hematopoietic Stem Cell Transplantation with Post-Transplant High-Dose Cyclophosphamide for Children and Adolescents with Hematologic Malignancies: An AIEOP-GITMO Retrospective Multicenter Study. Biol Blood Marrow Transplant 22 (5): 902-9, 2016. [PUBMED Abstract]
- Baker M, Wang H, Rowley SD, et al.: Comparative Outcomes after Haploidentical or Unrelated Donor Bone Marrow or Blood Stem Cell Transplantation in Adult Patients with Hematological Malignancies. Biol Blood Marrow Transplant 22 (11): 2047-2055, 2016. [PUBMED Abstract]
- Rashidi A, Slade M, DiPersio JF, et al.: Post-transplant high-dose cyclophosphamide after HLA-matched vs haploidentical hematopoietic cell transplantation for AML. Bone Marrow Transplant 51 (12): 1561-1564, 2016. [PUBMED Abstract]
- Fuchs EJ, O’Donnell PV, Eapen M, et al.: Double unrelated umbilical cord blood vs HLA-haploidentical bone marrow transplantation: the BMT CTN 1101 trial. Blood 137 (3): 420-428, 2021. [PUBMED Abstract]
- Symons HJ, Zahurak M, Cao Y, et al.: Myeloablative haploidentical BMT with posttransplant cyclophosphamide for hematologic malignancies in children and adults. Blood Adv 4 (16): 3913-3925, 2020. [PUBMED Abstract]
- Ciurea SO, Cao K, Fernandez-Vina M, et al.: The European Society for Blood and Marrow Transplantation (EBMT) Consensus Guidelines for the Detection and Treatment of Donor-specific Anti-HLA Antibodies (DSA) in Haploidentical Hematopoietic Cell Transplantation. Bone Marrow Transplant 53 (5): 521-534, 2018. [PUBMED Abstract]
- Bensinger WI, Martin PJ, Storer B, et al.: Transplantation of bone marrow as compared with peripheral-blood cells from HLA-identical relatives in patients with hematologic cancers. N Engl J Med 344 (3): 175-81, 2001. [PUBMED Abstract]
- Rocha V, Cornish J, Sievers EL, et al.: Comparison of outcomes of unrelated bone marrow and umbilical cord blood transplants in children with acute leukemia. Blood 97 (10): 2962-71, 2001. [PUBMED Abstract]
- Bertaina A, Merli P, Rutella S, et al.: HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood 124 (5): 822-6, 2014. [PUBMED Abstract]
- Eapen M, Horowitz MM, Klein JP, et al.: Higher mortality after allogeneic peripheral-blood transplantation compared with bone marrow in children and adolescents: the Histocompatibility and Alternate Stem Cell Source Working Committee of the International Bone Marrow Transplant Registry. J Clin Oncol 22 (24): 4872-80, 2004. [PUBMED Abstract]
- Shinzato A, Tabuchi K, Atsuta Y, et al.: PBSCT is associated with poorer survival and increased chronic GvHD than BMT in Japanese paediatric patients with acute leukaemia and an HLA-matched sibling donor. Pediatr Blood Cancer 60 (9): 1513-9, 2013. [PUBMED Abstract]
- Anasetti C, Logan BR, Lee SJ, et al.: Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med 367 (16): 1487-96, 2012. [PUBMED Abstract]
- Shimosato Y, Tanoshima R, Tsujimoto SI, et al.: Allogeneic Bone Marrow Transplantation versus Peripheral Blood Stem Cell Transplantation for Hematologic Malignancies in Children: A Systematic Review and Meta-Analysis. Biol Blood Marrow Transplant 26 (1): 88-93, 2020. [PUBMED Abstract]
- Milano F, Gooley T, Wood B, et al.: Cord-Blood Transplantation in Patients with Minimal Residual Disease. N Engl J Med 375 (10): 944-53, 2016. [PUBMED Abstract]
- Ruggeri A, Michel G, Dalle JH, et al.: Impact of pretransplant minimal residual disease after cord blood transplantation for childhood acute lymphoblastic leukemia in remission: an Eurocord, PDWP-EBMT analysis. Leukemia 26 (12): 2455-61, 2012. [PUBMED Abstract]
- Bachanova V, Burke MJ, Yohe S, et al.: Unrelated cord blood transplantation in adult and pediatric acute lymphoblastic leukemia: effect of minimal residual disease on relapse and survival. Biol Blood Marrow Transplant 18 (6): 963-8, 2012. [PUBMED Abstract]
- Sutton R, Shaw PJ, Venn NC, et al.: Persistent MRD before and after allogeneic BMT predicts relapse in children with acute lymphoblastic leukaemia. Br J Haematol 168 (3): 395-404, 2015. [PUBMED Abstract]
- Sanchez-Garcia J, Serrano J, Serrano-Lopez J, et al.: Quantification of minimal residual disease levels by flow cytometry at time of transplant predicts outcome after myeloablative allogeneic transplantation in ALL. Bone Marrow Transplant 48 (3): 396-402, 2013. [PUBMED Abstract]
- Mehta RS, Holtan SG, Wang T, et al.: GRFS and CRFS in alternative donor hematopoietic cell transplantation for pediatric patients with acute leukemia. Blood Adv 3 (9): 1441-1449, 2019. [PUBMED Abstract]
- Pulsipher MA, Chitphakdithai P, Logan BR, et al.: Donor, recipient, and transplant characteristics as risk factors after unrelated donor PBSC transplantation: beneficial effects of higher CD34+ cell dose. Blood 114 (13): 2606-16, 2009. [PUBMED Abstract]
- Aversa F, Terenzi A, Tabilio A, et al.: Full haplotype-mismatched hematopoietic stem-cell transplantation: a phase II study in patients with acute leukemia at high risk of relapse. J Clin Oncol 23 (15): 3447-54, 2005. [PUBMED Abstract]
- Kollman C, Howe CW, Anasetti C, et al.: Donor characteristics as risk factors in recipients after transplantation of bone marrow from unrelated donors: the effect of donor age. Blood 98 (7): 2043-51, 2001. [PUBMED Abstract]
- Kollman C, Spellman SR, Zhang MJ, et al.: The effect of donor characteristics on survival after unrelated donor transplantation for hematologic malignancy. Blood 127 (2): 260-7, 2016. [PUBMED Abstract]
- Boeckh M, Nichols WG: The impact of cytomegalovirus serostatus of donor and recipient before hematopoietic stem cell transplantation in the era of antiviral prophylaxis and preemptive therapy. Blood 103 (6): 2003-8, 2004. [PUBMED Abstract]
- Seebach JD, Stussi G, Passweg JR, et al.: ABO blood group barrier in allogeneic bone marrow transplantation revisited. Biol Blood Marrow Transplant 11 (12): 1006-13, 2005. [PUBMED Abstract]
- Logan AC, Wang Z, Alimoghaddam K, et al.: ABO mismatch is associated with increased nonrelapse mortality after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 21 (4): 746-54, 2015. [PUBMED Abstract]
- Stussi G, Muntwyler J, Passweg JR, et al.: Consequences of ABO incompatibility in allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 30 (2): 87-93, 2002. [PUBMED Abstract]
- Loren AW, Bunin GR, Boudreau C, et al.: Impact of donor and recipient sex and parity on outcomes of HLA-identical sibling allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 12 (7): 758-69, 2006. [PUBMED Abstract]
- Canaani J, Savani BN, Labopin M, et al.: ABO incompatibility in mismatched unrelated donor allogeneic hematopoietic cell transplantation for acute myeloid leukemia: A report from the acute leukemia working party of the EBMT. Am J Hematol 92 (8): 789-796, 2017. [PUBMED Abstract]
- Shaw BE, Logan BR, Spellman SR, et al.: Development of an Unrelated Donor Selection Score Predictive of Survival after HCT: Donor Age Matters Most. Biol Blood Marrow Transplant 24 (5): 1049-1056, 2018. [PUBMED Abstract]
- Stern M, Ruggeri L, Mancusi A, et al.: Survival after T cell-depleted haploidentical stem cell transplantation is improved using the mother as donor. Blood 112 (7): 2990-5, 2008. [PUBMED Abstract]
- Ciurea SO, Champlin RE: Donor selection in T cell-replete haploidentical hematopoietic stem cell transplantation: knowns, unknowns, and controversies. Biol Blood Marrow Transplant 19 (2): 180-4, 2013. [PUBMED Abstract]
- Wang Y, Chang YJ, Xu LP, et al.: Who is the best donor for a related HLA haplotype-mismatched transplant? Blood 124 (6): 843-50, 2014. [PUBMED Abstract]
- Canaani J, Savani BN, Labopin M, et al.: Impact of ABO incompatibility on patients’ outcome after haploidentical hematopoietic stem cell transplantation for acute myeloid leukemia – a report from the Acute Leukemia Working Party of the EBMT. Haematologica 102 (6): 1066-1074, 2017. [PUBMED Abstract]
- DeZern AE, Franklin C, Tsai HL, et al.: Relationship of donor age and relationship to outcomes of haploidentical transplantation with posttransplant cyclophosphamide. Blood Adv 5 (5): 1360-1368, 2021. [PUBMED Abstract]
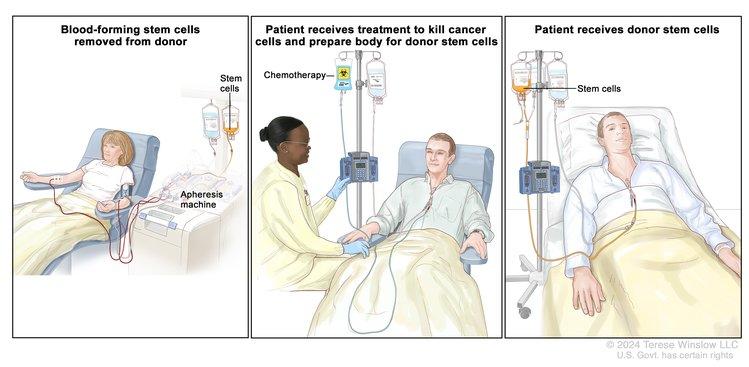
Allogeneic Hematopoietic Stem Cell Transplant (HSCT) Preparative Regimens
In the days just before infusion of the stem cell product (bone marrow, peripheral blood stem cells [PBSCs], or cord blood), HSCT recipients receive chemotherapy/immunotherapy, sometimes combined with radiation therapy. This is called a preparative regimen, and the original intent of this treatment was to:
- Create bone marrow space in the recipient for the donor cells to engraft.
- Suppress the immune system or eliminate the recipient T cells to minimize risk of rejection.
- Intensely treat cancer (if present) with high doses of active agents to overcome therapy resistance.
With the recognition that donor T cells can facilitate engraftment and kill tumors through graft-versus-leukemia (GVL) effects (obviating the need to create bone marrow space and intensely treat cancer), reduced-intensity or minimal-intensity HSCT approaches focusing on immune suppression rather than myeloablation have been developed. The resulting lower toxicity associated with these regimens has led to lower rates of transplant-related mortality and expanded eligibility for allogeneic HSCT to older individuals and younger patients with pre-HSCT comorbidities that put them at risk of severe toxicity after standard HSCT approaches.[1]
Existing preparative regimens vary tremendously in the amount of immunosuppression and myelosuppression they cause, with the lowest-intensity regimens relying heavily on a strong graft-versus-tumor (GVT) effect (see Figure 3).

Although these regimens lead to varying degrees of myelosuppression and immune suppression, they have been grouped clinically into the following three major categories (see Figure 4):[2]
- Myeloablative: Intense approaches that cause irreversible pancytopenia that requires stem cell rescue for restoration of hematopoiesis.
- Nonmyeloablative: Regimens that cause minimal cytopenias and do not require stem cell support.
- Reduced-intensity conditioning: Regimens that are of intermediate intensity and do not meet the definitions of nonmyeloablative or myeloablative regimens.

For years, retrospective studies showed similar outcomes using reduced-intensity and myeloablative approaches.[3,4] However, a Blood and Marrow Transplant Clinical Trials Network trial of adult patients with acute myeloid leukemia (AML) and myelodysplastic neoplasms (MDS) who were randomly assigned to receive either myeloablative or reduced-intensity HSCT approaches demonstrated the importance of regimen intensity.[5]
- At 18 months, relapse was markedly higher in the reduced-intensity cohort (48% vs. 13.5%, P < .001).
- Although treatment-related mortality was higher in the myeloablative arm (16% vs. 4%, P = .002), relapse-free survival rates were superior (69% vs. 47%, P < .01) and overall survival (OS) rates were higher in the myeloablative arm (76% vs. 68%), with a nonsignificant P value of .07.
With this in mind, the use of reduced-intensity conditioning and nonmyeloablative regimens is well established in older adults who cannot tolerate more intense myeloablative approaches,[6–8] but these approaches have been studied in a limited number of younger patients with malignancies.[9–13] A large Pediatric Blood and Marrow Transplant Consortium study identified patients at high risk of transplant-related mortality with myeloablative regimens (e.g., history of previous myeloablative transplant, severe organ system dysfunction, or active, invasive fungal infection) and successfully treated these patients with a reduced-intensity regimen.[14] Transplant-related mortality was low in this high-risk group, and long-term survival occurred in most patients with minimal or no detectable disease at the time of transplant. Because the risks of relapse are higher with these approaches, their use in pediatric cancer is currently limited to patients ineligible for myeloablative regimens and is most likely to be successful when patients have achieved minimal residual disease (MRD)–negative remissions.[14]
In pediatric HSCT, there has been an effort to develop preparative regimens that are myeloablative but do not have the severe toxicities associated with traditional, highly intense myeloablative approaches such as full-dose total-body irradiation, busulfan and cyclophosphamide, or busulfan, cyclophosphamide, and melphalan. These less-intense regimens are called reduced toxicity and include approaches such as full-dose busulfan and fludarabine or treosulfan and fludarabine. These approaches have been especially useful in transplant for nonmalignant disorders that require full chimerism,[15] but they have often shown similar outcomes when used for patients with malignancies.[16]
Establishing Donor Chimerism
Intense myeloablative approaches almost invariably result in hematopoiesis derived from donor cells upon count recovery after the transplant. The introduction of reduced-toxicity, reduced-intensity, and nonmyeloablative conditioning into HSCT practice has resulted in a slower pace of transition to donor hematopoiesis (gradually increasing from partial to full donor hematopoiesis over months) that sometimes remains partially long-term. DNA-based techniques have been established to differentiate donor from recipient hematopoiesis, applying the word chimerism to describe whether all or part of hematopoiesis after HSCT is from the donor or recipient.
There are several implications regarding the pace and extent of donor chimerism achieved by an HSCT recipient. For patients receiving reduced-intensity conditioning or nonmyeloablative regimens, rapid progression to full donor chimerism is associated with lower relapse rates but more graft-versus-host disease (GVHD).[17] The delayed pace of obtaining full donor chimerism after reduced-intensity regimens has led to late-onset acute GVHD, occurring as late as 6 to 7 months after HSCT (acute GVHD generally occurs within 100 days after myeloablative approaches).[18] A portion of patients achieve stable mixed chimerism of both donor and recipient. Mixed chimerism is associated with more relapse after HSCT for malignancies and less GVHD. However, this condition is often advantageous for nonmalignant HSCT, where usually only a percentage of normal hematopoiesis is needed to correct the underlying disorder and GVHD is not beneficial.[19] Finally, serially measured decreasing donor chimerism, especially T-cell–specific chimerism, has been associated with increased risk of rejection.[20]
Because of the implications of persistent recipient chimerism, most transplant programs test for chimerism shortly after engraftment and continue testing regularly until stable full donor hematopoiesis has been achieved. Investigators have defined two approaches to treat the increased risks of relapse and rejection associated with increasing recipient chimerism: rapid withdrawal of immune suppression and donor lymphocyte infusions (DLI). These approaches are frequently used to address this issue, and they have been shown to decrease relapse risk and stop rejection in some cases.[21–23] The timing of immune suppression and dose tapers and approaches to administration of DLI to increase or stabilize donor chimerism vary between stem cell sources. There is also a wide institutional variability, with some institutions proactively following chimerism and often intervening, and others having a more limited approach to interventions. For more information, see the Using donor lymphocyte infusions (DLI) or early withdrawal of immune suppression to enhance GVL section.
References
- Deeg HJ, Sandmaier BM: Who is fit for allogeneic transplantation? Blood 116 (23): 4762-70, 2010. [PUBMED Abstract]
- Bacigalupo A, Ballen K, Rizzo D, et al.: Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant 15 (12): 1628-33, 2009. [PUBMED Abstract]
- Luger SM, Ringdén O, Zhang MJ, et al.: Similar outcomes using myeloablative vs reduced-intensity allogeneic transplant preparative regimens for AML or MDS. Bone Marrow Transplant 47 (2): 203-11, 2012. [PUBMED Abstract]
- Pulsipher MA, Chitphakdithai P, Logan BR, et al.: Donor, recipient, and transplant characteristics as risk factors after unrelated donor PBSC transplantation: beneficial effects of higher CD34+ cell dose. Blood 114 (13): 2606-16, 2009. [PUBMED Abstract]
- Scott BL, Pasquini MC, Logan BR, et al.: Myeloablative Versus Reduced-Intensity Hematopoietic Cell Transplantation for Acute Myeloid Leukemia and Myelodysplastic Syndromes. J Clin Oncol 35 (11): 1154-1161, 2017. [PUBMED Abstract]
- Giralt S, Estey E, Albitar M, et al.: Engraftment of allogeneic hematopoietic progenitor cells with purine analog-containing chemotherapy: harnessing graft-versus-leukemia without myeloablative therapy. Blood 89 (12): 4531-6, 1997. [PUBMED Abstract]
- Slavin S, Nagler A, Naparstek E, et al.: Nonmyeloablative stem cell transplantation and cell therapy as an alternative to conventional bone marrow transplantation with lethal cytoreduction for the treatment of malignant and nonmalignant hematologic diseases. Blood 91 (3): 756-63, 1998. [PUBMED Abstract]
- Storb R, Yu C, Sandmaier BM, et al.: Mixed hematopoietic chimerism after marrow allografts. Transplantation in the ambulatory care setting. Ann N Y Acad Sci 872: 372-5; discussion 375-6, 1999. [PUBMED Abstract]
- Bradley MB, Satwani P, Baldinger L, et al.: Reduced intensity allogeneic umbilical cord blood transplantation in children and adolescent recipients with malignant and non-malignant diseases. Bone Marrow Transplant 40 (7): 621-31, 2007. [PUBMED Abstract]
- Del Toro G, Satwani P, Harrison L, et al.: A pilot study of reduced intensity conditioning and allogeneic stem cell transplantation from unrelated cord blood and matched family donors in children and adolescent recipients. Bone Marrow Transplant 33 (6): 613-22, 2004. [PUBMED Abstract]
- Gómez-Almaguer D, Ruiz-Argüelles GJ, Tarín-Arzaga Ldel C, et al.: Reduced-intensity stem cell transplantation in children and adolescents: the Mexican experience. Biol Blood Marrow Transplant 9 (3): 157-61, 2003. [PUBMED Abstract]
- Pulsipher MA, Woolfrey A: Nonmyeloablative transplantation in children. Current status and future prospects. Hematol Oncol Clin North Am 15 (5): 809-34, vii-viii, 2001. [PUBMED Abstract]
- Roman E, Cooney E, Harrison L, et al.: Preliminary results of the safety of immunotherapy with gemtuzumab ozogamicin following reduced intensity allogeneic stem cell transplant in children with CD33+ acute myeloid leukemia. Clin Cancer Res 11 (19 Pt 2): 7164s-7170s, 2005. [PUBMED Abstract]
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009. [PUBMED Abstract]
- Cseh A, Galimard JE, de la Fuente J, et al.: Busulfan-fludarabine- or treosulfan-fludarabine-based conditioning before allogeneic HSCT from matched sibling donors in paediatric patients with sickle cell disease: A study on behalf of the EBMT Paediatric Diseases and Inborn Errors Working Parties. Br J Haematol 204 (1): e1-e5, 2024. [PUBMED Abstract]
- Pulsipher MA, Ahn KW, Bunin NJ, et al.: KIR-favorable TCR-αβ/CD19-depleted haploidentical HCT in children with ALL/AML/MDS: primary analysis of the PTCTC ONC1401 trial. Blood 140 (24): 2556-2572, 2022. [PUBMED Abstract]
- Baron F, Baker JE, Storb R, et al.: Kinetics of engraftment in patients with hematologic malignancies given allogeneic hematopoietic cell transplantation after nonmyeloablative conditioning. Blood 104 (8): 2254-62, 2004. [PUBMED Abstract]
- Vigorito AC, Campregher PV, Storer BE, et al.: Evaluation of NIH consensus criteria for classification of late acute and chronic GVHD. Blood 114 (3): 702-8, 2009. [PUBMED Abstract]
- Marsh RA, Vaughn G, Kim MO, et al.: Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood 116 (26): 5824-31, 2010. [PUBMED Abstract]
- McSweeney PA, Niederwieser D, Shizuru JA, et al.: Hematopoietic cell transplantation in older patients with hematologic malignancies: replacing high-dose cytotoxic therapy with graft-versus-tumor effects. Blood 97 (11): 3390-400, 2001. [PUBMED Abstract]
- Bader P, Kreyenberg H, Hoelle W, et al.: Increasing mixed chimerism is an important prognostic factor for unfavorable outcome in children with acute lymphoblastic leukemia after allogeneic stem-cell transplantation: possible role for pre-emptive immunotherapy? J Clin Oncol 22 (9): 1696-705, 2004. [PUBMED Abstract]
- Horn B, Soni S, Khan S, et al.: Feasibility study of preemptive withdrawal of immunosuppression based on chimerism testing in children undergoing myeloablative allogeneic transplantation for hematologic malignancies. Bone Marrow Transplant 43 (6): 469-76, 2009. [PUBMED Abstract]
- Haines HL, Bleesing JJ, Davies SM, et al.: Outcomes of donor lymphocyte infusion for treatment of mixed donor chimerism after a reduced-intensity preparative regimen for pediatric patients with nonmalignant diseases. Biol Blood Marrow Transplant 21 (2): 288-92, 2015. [PUBMED Abstract]
Immunotherapeutic Effects of Allogeneic Hematopoietic Stem Cell Transplant (HSCT)
Graft-Versus-Leukemia (GVL) Effect
Early studies in HSCT focused on the delivery of intense myeloablative preparative regimens followed by rescue of the hematopoietic system with either an autologous or allogeneic bone marrow transplant. Investigators quickly showed that allogeneic approaches led to a decreased risk of relapse caused by an immunotherapeutic reaction of the new bone marrow graft against tumor antigens. This phenomenon came to be termed the GVL or graft-versus-tumor (GVT) effect and has been associated with mismatches to both major and minor HLA antigens.
The GVL effect is challenging to use therapeutically because of a strong association between GVL and clinical graft-versus-host disease (GVHD). For standard approaches to HSCT, the highest survival rates have been associated with mild or moderate GVHD (grades I to II in acute myeloid leukemia [AML] and grades I to III in acute lymphoblastic leukemia [ALL]), compared with patients who have no GVHD and experience more relapse or patients with severe GVHD who experience more transplant-related mortality.[1–3]; [4][Level of evidence C2]
Understanding when GVL occurs and how to use GVL optimally is challenging. One method of study compares rates of relapse and survival between patients undergoing myeloablative HSCT with either autologous or allogeneic donors for a given disease.
- Leukemia and myelodysplastic neoplasms (MDS): A clear advantage has been noted when allogeneic approaches are used for ALL, AML, chronic myelogenous leukemia (CML), and MDS. For ALL and AML specifically, autologous HSCT approaches for most high-risk patient groups have shown results similar to those obtained with chemotherapy, while allogeneic approaches produced superior results and are therefore useful for chemoresistant or relapsed patients.[5,6]
- Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL): Patients with HL or NHL generally fare better with autologous approaches, although there may be a role for allogeneic approaches in relapsed lymphoblastic lymphoma, lymphoma that is poorly responsive to chemotherapy, or lymphoma that has relapsed after autologous HSCT.[7]
Further insights into the therapeutic benefit of GVL/GVT for given diseases have come from the use of reduced-intensity preparative regimens. This approach to transplant relies on GVL because, in most cases, the intensity of the preparative regimen is not sufficient for cure. Although studies have shown benefit for patients pursuing this approach when they are ineligible for standard transplant,[8] this approach has not been used for most children with cancer who require HSCT because pediatric cancer patients can generally undergo myeloablative approaches safely. For more information, see the Allogeneic HSCT Preparative Regimens section.
Using donor lymphocyte infusions (DLI) or early withdrawal of immune suppression to enhance GVL
GVL can be achieved therapeutically through the infusion of cells after transplant that either specifically or nonspecifically target the tumor. The most common approach is the use of DLI. This approach relies on the persistence of donor T-cell engraftment after transplant to prevent rejection of donor lymphocytes infused to induce GVL.
Therapeutic DLI results in potent responses in patients with CML who relapse after transplant (60%–80% enter into long-term remission),[9] but responses in patients with other diseases (such as AML and ALL) have been less potent, with long-term survival rates of only 20% to 30%.[10] DLI works poorly in patients with acute leukemia who relapse early and who have high levels of active disease. Late relapse (>6 months after transplant) and the treatment of patients into complete remission with chemotherapy before DLI have been associated with improved outcomes.[11] Infusions of donor lymphocytes modified to enhance GVL or other donor cells (e.g., natural killer [NK] cells) have also been studied but have yet to be generally adopted.
Another method of delivering GVL therapeutically is the rapid withdrawal of immune suppression after HSCT. Some studies have scheduled more rapid immune suppression tapers based on donor type (related donors are tapered more quickly than are unrelated donors because of less GVHD risk), and others have used sensitive measures of either low levels of persistent recipient cells (recipient chimerism) or minimal residual disease to assess the risk of relapse and trigger rapid taper of immune suppression.
A combination of early withdrawal of immune suppression after HSCT with DLI to prevent relapse in patients at high risk of relapse because of persistent/progressive recipient chimerism has been tested in patients who underwent transplant for both ALL and AML.[12][Level of evidence B4]; [13][Level of evidence C2]
- ALL: One study found increasing recipient chimerism in 46 of 101 patients with ALL. Thirty-one of those patients had withdrawal of immune suppression, and a portion went on to receive DLI if GVHD did not occur. This group had a survival rate of 37%, compared with 0% in the 15 patients who did not undergo this approach (P < .001).[14]
- AML: About 20% of patients with AML experienced mixed chimerism after HSCT and were identified as high risk. Of these patients, 54% survived if they underwent withdrawal of immune suppression with or without DLI. There were no survivors among those who did not receive this therapy.[15]
Other immunological and cell therapy approaches under evaluation
Role of killer immunoglobulin-like receptor (KIR) mismatching in HSCT
Donor-derived NK cells in the post-HSCT setting have been shown to promote the following:[16–18]
- Engraftment.
- Decreased GVHD.
- Fewer relapses of hematological malignancies.
- Improved survival.
NK-cell function is modulated by interactions with a number of receptor families, including activating and inhibiting KIR. The KIR effect in the allogeneic HSCT setting hinges on the expression of specific inhibitory KIR on donor-derived NK cells and either the presence or absence of their matching HLA class I molecules (KIR ligands) on recipient leukemic and normal cells. Normally, the presence of specific KIR ligands interacting with paired inhibitory KIR molecules prevents NK cell attacks on healthy cells. In the allogeneic transplant setting, recipient leukemia cells genetically differ from donor NK cells, and they may not have the appropriate inhibitory KIR ligand. Mismatch of ligand and receptor allows NK-cell–based killing of recipient leukemia cells to proceed for certain donor-recipient genetic combinations.
The original observation of decreased relapse with certain KIR-ligand combinations was made in the setting of T-cell–depleted haploidentical transplant and was strongest after HSCT for AML.[17,19] However, some haploidentical transplant studies have not shown this effect.[20] Along with decreasing relapse, these studies have suggested a decrease in GVHD with appropriate KIR-ligand combinations. Many subsequent studies did not detect survival effects for KIR-incompatible HSCT using standard transplant methods,[21–24] which has led to the conclusion that T-cell depletion may be necessary to remove other forms of inhibitory cellular interactions.
Decreased relapse and better survival have been noted with donor/recipient KIR-ligand incompatibility after cord blood HSCT, a relatively T-cell–depleted procedure.[25,26] In contrast to this notion, one study demonstrated that some KIR mismatching combinations (activating receptor KIR2DS1 with the HLA C1 ligand) can lead to decreased relapse after AML HSCT without T-cell depletion.[27] The role of KIR incompatibility in sibling donor HSCT and in diseases other than AML is controversial, but in pediatrics, at least two groups have found better outcomes with specific types of KIR mismatching in ALL.[28–30]
A current challenge associated with studies of KIR is that several different approaches have been used to determine what is KIR incompatible and what are the most favorable combinations of KIR molecules in donor-recipient pairs.[19,31,32] Activating KIR molecules have also been shown to contribute to the effect.[33] The standardization of KIR classification and prospective studies should help clarify the utility and importance of this approach. Because a limited number of centers perform haploidentical HSCT and the results of other approaches to HSCT are preliminary, most transplant programs do not use KIR mismatching as part of their strategy for choosing a donor. Full HLA matching is considered most important for outcome, with considerations of KIR mismatching or choosing donors with favorable KIR activation profiles remaining secondary.
NK-cell transplant
With a low risk of GVHD and demonstrated efficacy in decreasing relapse in posthaploidentical HSCT settings, NK-cell infusions as a method of treating high-risk patients and consolidating patients in remission have been studied:
Evidence (NK-cell transplant outcomes):
- A University of Minnesota research group compared approaches with different NK-cell populations.[34]
- The study initially failed to demonstrate efficacy with autologous NK cells, but it found that intense immunoablative therapy followed by purified haploidentical NK cells and interleukin-2 (IL-2) maintenance led to remission in 5 of 19 high-risk patients with AML.
- Researchers at St. Jude Children’s Research Hospital treated ten intermediate-risk patients with AML who had completed chemotherapy and were in remission. The patients received lower-dose immunosuppression followed by haploidentical NK-cell infusions and IL-2 for consolidation.[35]
- Expansion of NK cells was noted in all nine of the KIR-incompatible donor-recipient pairs.
- All ten children remained in remission at 2 years.
- A follow-up phase II study is under way, as are many investigations into NK-cell therapy for a number of cancer types.
Although early survival rates are high in this high-risk AML cohort, multicenter confirmatory studies will be necessary to establish the efficacy of these types of NK-cell approaches.
- Other investigators have used expanded/activated NK cells before and after HSCT.[36] One approach that included the culturing of haploidentical NK cells with membrane-bound IL-21 showed marked expansion and high activity. These cells were then infused just before haploidentical HSCT, followed by additional infusions on day 7 and 28 after HSCT.[36]
References
- Yeshurun M, Weisdorf D, Rowe JM, et al.: The impact of the graft-versus-leukemia effect on survival in acute lymphoblastic leukemia. Blood Adv 3 (4): 670-680, 2019. [PUBMED Abstract]
- Pulsipher MA, Langholz B, Wall DA, et al.: The addition of sirolimus to tacrolimus/methotrexate GVHD prophylaxis in children with ALL: a phase 3 Children’s Oncology Group/Pediatric Blood and Marrow Transplant Consortium trial. Blood 123 (13): 2017-25, 2014. [PUBMED Abstract]
- Neudorf S, Sanders J, Kobrinsky N, et al.: Allogeneic bone marrow transplantation for children with acute myelocytic leukemia in first remission demonstrates a role for graft versus leukemia in the maintenance of disease-free survival. Blood 103 (10): 3655-61, 2004. [PUBMED Abstract]
- Boyiadzis M, Arora M, Klein JP, et al.: Impact of Chronic Graft-versus-Host Disease on Late Relapse and Survival on 7,489 Patients after Myeloablative Allogeneic Hematopoietic Cell Transplantation for Leukemia. Clin Cancer Res 21 (9): 2020-8, 2015. [PUBMED Abstract]
- Woods WG, Neudorf S, Gold S, et al.: A comparison of allogeneic bone marrow transplantation, autologous bone marrow transplantation, and aggressive chemotherapy in children with acute myeloid leukemia in remission. Blood 97 (1): 56-62, 2001. [PUBMED Abstract]
- Ribera JM, Ortega JJ, Oriol A, et al.: Comparison of intensive chemotherapy, allogeneic, or autologous stem-cell transplantation as postremission treatment for children with very high risk acute lymphoblastic leukemia: PETHEMA ALL-93 Trial. J Clin Oncol 25 (1): 16-24, 2007. [PUBMED Abstract]
- Gross TG, Hale GA, He W, et al.: Hematopoietic stem cell transplantation for refractory or recurrent non-Hodgkin lymphoma in children and adolescents. Biol Blood Marrow Transplant 16 (2): 223-30, 2010. [PUBMED Abstract]
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009. [PUBMED Abstract]
- Porter DL, Collins RH, Shpilberg O, et al.: Long-term follow-up of patients who achieved complete remission after donor leukocyte infusions. Biol Blood Marrow Transplant 5 (4): 253-61, 1999. [PUBMED Abstract]
- Levine JE, Barrett AJ, Zhang MJ, et al.: Donor leukocyte infusions to treat hematologic malignancy relapse following allo-SCT in a pediatric population. Bone Marrow Transplant 42 (3): 201-5, 2008. [PUBMED Abstract]
- Warlick ED, DeFor T, Blazar BR, et al.: Successful remission rates and survival after lymphodepleting chemotherapy and donor lymphocyte infusion for relapsed hematologic malignancies postallogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 18 (3): 480-6, 2012. [PUBMED Abstract]
- Horn B, Petrovic A, Wahlstrom J, et al.: Chimerism-based pre-emptive immunotherapy with fast withdrawal of immunosuppression and donor lymphocyte infusions after allogeneic stem cell transplantation for pediatric hematologic malignancies. Biol Blood Marrow Transplant 21 (4): 729-37, 2015. [PUBMED Abstract]
- Horn B, Wahlstrom JT, Melton A, et al.: Early mixed chimerism-based preemptive immunotherapy in children undergoing allogeneic hematopoietic stem cell transplantation for acute leukemia. Pediatr Blood Cancer 64 (8): , 2017. [PUBMED Abstract]
- Bader P, Kreyenberg H, Hoelle W, et al.: Increasing mixed chimerism is an important prognostic factor for unfavorable outcome in children with acute lymphoblastic leukemia after allogeneic stem-cell transplantation: possible role for pre-emptive immunotherapy? J Clin Oncol 22 (9): 1696-705, 2004. [PUBMED Abstract]
- Rettinger E, Willasch AM, Kreyenberg H, et al.: Preemptive immunotherapy in childhood acute myeloid leukemia for patients showing evidence of mixed chimerism after allogeneic stem cell transplantation. Blood 118 (20): 5681-8, 2011. [PUBMED Abstract]
- Ruggeri L, Capanni M, Urbani E, et al.: Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 295 (5562): 2097-100, 2002. [PUBMED Abstract]
- Giebel S, Locatelli F, Lamparelli T, et al.: Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood 102 (3): 814-9, 2003. [PUBMED Abstract]
- Bari R, Rujkijyanont P, Sullivan E, et al.: Effect of donor KIR2DL1 allelic polymorphism on the outcome of pediatric allogeneic hematopoietic stem-cell transplantation. J Clin Oncol 31 (30): 3782-90, 2013. [PUBMED Abstract]
- Ruggeri L, Mancusi A, Capanni M, et al.: Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood 110 (1): 433-40, 2007. [PUBMED Abstract]
- Merli P, Algeri M, Galaverna F, et al.: TCRαβ/CD19 cell-depleted HLA-haploidentical transplantation to treat pediatric acute leukemia: updated final analysis. Blood 143 (3): 279-289, 2024. [PUBMED Abstract]
- Davies SM, Ruggieri L, DeFor T, et al.: Evaluation of KIR ligand incompatibility in mismatched unrelated donor hematopoietic transplants. Killer immunoglobulin-like receptor. Blood 100 (10): 3825-7, 2002. [PUBMED Abstract]
- Farag SS, Bacigalupo A, Eapen M, et al.: The effect of KIR ligand incompatibility on the outcome of unrelated donor transplantation: a report from the center for international blood and marrow transplant research, the European blood and marrow transplant registry, and the Dutch registry. Biol Blood Marrow Transplant 12 (8): 876-84, 2006. [PUBMED Abstract]
- Davies SM, Iannone R, Alonzo TA, et al.: A Phase 2 Trial of KIR-Mismatched Unrelated Donor Transplantation Using in Vivo T Cell Depletion with Antithymocyte Globulin in Acute Myelogenous Leukemia: Children’s Oncology Group AAML05P1 Study. Biol Blood Marrow Transplant 26 (4): 712-717, 2020. [PUBMED Abstract]
- Verneris MR, Miller JS, Hsu KC, et al.: Investigation of donor KIR content and matching in children undergoing hematopoietic cell transplantation for acute leukemia. Blood Adv 4 (7): 1350-1356, 2020. [PUBMED Abstract]
- Cooley S, Trachtenberg E, Bergemann TL, et al.: Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood 113 (3): 726-32, 2009. [PUBMED Abstract]
- Willemze R, Rodrigues CA, Labopin M, et al.: KIR-ligand incompatibility in the graft-versus-host direction improves outcomes after umbilical cord blood transplantation for acute leukemia. Leukemia 23 (3): 492-500, 2009. [PUBMED Abstract]
- Venstrom JM, Pittari G, Gooley TA, et al.: HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med 367 (9): 805-16, 2012. [PUBMED Abstract]
- Leung W: Use of NK cell activity in cure by transplant. Br J Haematol 155 (1): 14-29, 2011. [PUBMED Abstract]
- Leung W, Campana D, Yang J, et al.: High success rate of hematopoietic cell transplantation regardless of donor source in children with very high-risk leukemia. Blood 118 (2): 223-30, 2011. [PUBMED Abstract]
- Oevermann L, Michaelis SU, Mezger M, et al.: KIR B haplotype donors confer a reduced risk for relapse after haploidentical transplantation in children with ALL. Blood 124 (17): 2744-7, 2014. [PUBMED Abstract]
- Leung W, Iyengar R, Triplett B, et al.: Comparison of killer Ig-like receptor genotyping and phenotyping for selection of allogeneic blood stem cell donors. J Immunol 174 (10): 6540-5, 2005. [PUBMED Abstract]
- Pulsipher MA, Ahn KW, Bunin NJ, et al.: KIR-favorable TCR-αβ/CD19-depleted haploidentical HCT in children with ALL/AML/MDS: primary analysis of the PTCTC ONC1401 trial. Blood 140 (24): 2556-2572, 2022. [PUBMED Abstract]
- Cooley S, Weisdorf DJ, Guethlein LA, et al.: Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood 116 (14): 2411-9, 2010. [PUBMED Abstract]
- Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al.: Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 105 (8): 3051-7, 2005. [PUBMED Abstract]
- Rubnitz JE, Inaba H, Ribeiro RC, et al.: NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol 28 (6): 955-9, 2010. [PUBMED Abstract]
- Ciurea SO, Schafer JR, Bassett R, et al.: Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood 130 (16): 1857-1868, 2017. [PUBMED Abstract]
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ASCT2031 (NCT05457556) (Mismatched Related Donor [Haplo] Versus Matched Unrelated Donor [MUD] Hematopoietic Stem Cell Transplant [HSCT] for Children, Adolescents, and Young Adults with Acute Leukemia or Myelodysplastic Neoplasms [MDS]): This is a phase III randomized controlled trial that will compare outcomes between haplo HSCT and MUD HSCT for patients with acute leukemias or MDS. Patients with both available donor sources (MUD and haplo) will be randomly assigned to either Arm A (haplo HSCT) or Arm B (MUD HSCT). The randomization will be stratified by patient age, complete remission status, and disease type. Patients who do not have both an available MUD and haplo donor, but do have a haplo donor, will be assigned to Arm C and receive a nonrandomized haplo HSCT. Arm C is expected to enroll a higher fraction of racial and ethnic minority patients because they lack MUDs more often than White patients. This third arm will allow direct comparisons of haplo HSCT outcomes between racial and ethnic minority patients and White patients.
The clinically relevant end points, severe graft-versus-host disease (GVHD), and disease-free survival (DFS) in haplo HSCT versus MUD HSCT will be compared as co-primary objectives. The hypothesis is that the incidence of severe GVHD will be less frequent and DFS will be noninferior for haplo HSCT than MUD HSCT.
Latest Updates to This Summary (06/13/2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
This summary was comprehensively reviewed.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the use of allogeneic hematopoietic stem cell transplant in treating pediatric cancer. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Pediatric Allogeneic Hematopoietic Stem Cell Transplant are:
- Thomas G. Gross, MD, PhD (National Cancer Institute)
- Michael A. Pulsipher, MD (Huntsman Cancer Institute at University of Utah)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Pediatric Allogeneic Hematopoietic Stem Cell Transplant. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: /types/childhood-cancers/hp-stem-cell-transplant/allogeneic. Accessed <MM/DD/YYYY>. [PMID: 35133766]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
. There are also two pullouts: one showing a close up of the inside structure of a lymph node and the attached artery, vein, and lymph vessels with arrows showing how the lymph (clear, watery fluid) moves into and out of the lymph node, and another showing a close up of bone marrow with blood cells.")
; left panel shows blood being drawn from a vein on the inside of the elbow using a tube attached to a syringe; right panel shows a laboratory test tube with blood cells separated into layers: plasma, white blood cells, platelets, and red blood cells.")





 the top left panel shows cancer in the liver; (b) the top right panel shows cancer in the left lung and in two groups of lymph nodes below the diaphragm; (c) the bottom left panel shows cancer in the left lung and in a group of lymph nodes above the diaphragm and below the diaphragm; and (d) the bottom right panel shows cancer in both lungs, the liver, and the bone marrow (pullout). Also shown is primary cancer in the lymph nodes and a pullout of the brain with cerebrospinal fluid (in blue).")
 to PD-1 (on the T cell), which keeps T cells from killing tumor cells in the body. Also shown are a tumor cell antigen and T cell receptor. The panel on the right shows immune checkpoint inhibitors (anti-PD-L1 and anti-PD-1) blocking the binding of PD-L1 to PD-1, which allows the T cells to kill tumor cells.")
 moves into and out of the lymph node. Another inset shows a close up of bone marrow with blood cells.")
 scan; drawing shows a child lying on a table that slides through the CT scanner, which takes a series of detailed x-ray pictures of areas inside the body.")
 scan; drawing shows a child lying on table that slides through the PET scanner.")
 scan; drawing shows a child lying on a table that slides into the MRI machine, which takes a series of detailed pictures of areas inside the body.")



. An inset shows a lymph node with a lymph vessel, an artery, and a vein. Cancer cells are shown inside the lymph node.")
 cancer in lymph node groups above and below the diaphragm. Also shown is (b) cancer in a lymph node group above the diaphragm and cancer in the spleen. The lungs are also shown. An inset shows a close-up of a lymph node with a lymph vessel, an artery, and a vein. Cancer cells are shown inside the lymph node.")
 the top left panel shows cancer in the liver; (b) the top right panel shows cancer in the left lung and in two groups of lymph nodes below the diaphragm; (c) the bottom left panel shows cancer in the left lung and in a group of lymph nodes above the diaphragm and below the diaphragm; and (d) the bottom right panel shows cancer in both lungs and the liver. There is also a pullout showing cancer in the bone marrow.")
 being inserted into the lower back. Inset shows a close-up of the spinal needle inserted into the cerebrospinal fluid (CSF) in the lower part of the spinal column.")
 against the skin of the abdomen. A computer screen shows a sonogram (picture).")




 in the brain and spinal cord, and an Ommaya reservoir (a dome-shaped container that is placed under the scalp during surgery; it holds the drugs as they flow through a small tube into the brain). Top section shows a syringe and needle injecting anticancer drugs into the Ommaya reservoir. Bottom section shows a syringe and needle injecting anticancer drugs directly into the cerebrospinal fluid in the lower part of the spinal column.")
: Drawing of stem cells being collected from a donor's bloodstream using an apheresis machine. Blood is removed from a vein in the donor's arm and flows through the machine where the stem cells are removed. The rest of the blood is then returned to the donor through a vein in their other arm. (Panel 2): Drawing of a health care provider giving a patient an infusion of chemotherapy through a catheter in the patient's chest. The chemotherapy is given to kill cancer cells and prepare the patient's body for the donor stem cells. (Panel 3): Drawing of a patient receiving an infusion of the donor stem cells through a catheter in the chest.")
 being made in the laboratory; the gene for CAR is inserted into the T cells and then millions of CAR T cells are grown. Drawing also shows the CAR T cells being given to the patient by infusion and binding to antigens on the cancer cells and killing them.")