SUMMARY: The FDA on July 30, 2020, approved TECENTRIQ® (Atezolizumab), in combination with COTELLIC® (Cobimetinib) and ZELBORAF® (Vemurafenib), for patients with BRAF V600 mutation-positive unresectable or metastatic melanoma. It is estimated that in the US, approximately 100,350 new cases of melanoma will be diagnosed in 2020 and approximately 6,850 patients are expected to die of the disease. The incidence of melanoma has been on the rise for the past three decades. Surgical resection with a curative intent is the standard of care for patients with early stage melanoma, with a 5-year survival rate of 98% for Stage I disease and 90% for Stage II disease. Patients with locally advanced or metastatic melanoma historically have had poor outcomes. With the development and availability of immune checkpoint inhibitors and BRAF and MEK inhibitors, this patient group now has significantly improved outcomes.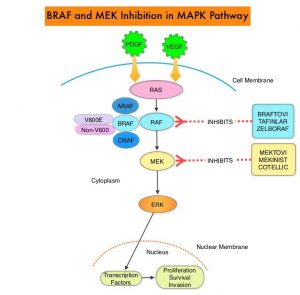
The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been detected in 6-8% of all malignancies. The most common BRAF mutation in melanoma is at the V600E/K site and is detected in approximately 50% of melanomas, and result in constitutive activation of the MAPK pathway.
ZELBORAF® (Vemurafenib), a selective oral inhibitor of mutated BRAF, demonstrated significant improvement in Progression Free Survival (PFS) and Overall Survival (OS), compared to Dacarbazine. Squamous cell carcinomas were seen in about 6% of the patients treated with BRAF inhibitors. Paradoxical activation of the MAPK pathway in cells without a BRAF mutation has been implicated in the emergence of drug resistance and increased incidence of BRAF-inhibitor induced skin tumors. MEK gene is downstream from RAF in the MAPK pathway. The addition of a selective inhibitor of MEK gene such as COTELLIC® (Cobimetinib) to a BRAF inhibitor such as ZELBORAF® has addressed some of these limitations, in previously published studies, with improvement in Objective Response Rates (ORR) and decrease in the incidence of cutaneous secondary cancers. coBRIM is a multicenter, randomized, Phase III study in which the efficacy and safety of COTELLIC® combined with ZELBORAF®, was evaluated in previously untreated patients, with advanced BRAF-mutated melanoma. The final analysis of this trial evaluated the 5-year survival data, and the OS was over 30% in patients who received the combination therapy, with a Complete Response (CR) rate was about 20%.
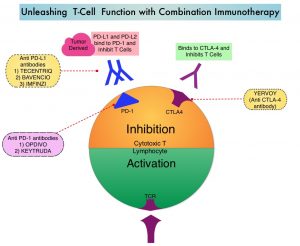
TECENTRIQ® (Atezolizumab) is an anti PD-L1 monoclonal antibody, designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors. PD-L1 inhibition may prevent T-cell deactivation and further enable the activation of T cells. The 5 year OS among patients receiving PD1 targeted immunotherapy is about 34%, with a median OS of 17-20 months. With the approval of multiple therapeutic options for the management of patients with BRAF-mutant melanoma, treatment decisions have become increasingly complex. In patients with limited disease burden, immunotherapy with checkpoint inhibitors is favored by most clinicians, based on the long term data supporting the durability of responses with immunotherapies, but response rates are lower. On the contrary, BRAF-targeted agents are utilized in patients with extensive, symptomatic disease and active brain metastases, as the response rates are higher but are short lived. The optimal sequence of these therapeutic strategies in order to improve long-term patient outcome, has remained unclear.
Preclinical studies suggested that combining these two targeted therapies with a checkpoint inhibitor might overcome the limitations of each class and potentially lead to more durable responses. The safety and efficacy of combining TECENTRIQ® with COTELLIC® (MEK inhibitor) and ZELBORAF® (BRAF inhibitor), in patients with BRAFV600-mutated metastatic melanoma, was evaluated in a Phase I study, with promising results, and a 28-day run-in period with COTELLIC® and ZELBORAF® was associated with an increase in proliferating CD4+ T-helper cells, without increasing the T-regulatory cells (Tregs). Tumor cells use Tregs as a shield to protect themselves against anti-tumor immune response and Tregs remain a hurdle in achieving the complete potential of anti-cancer therapies including immunotherapy. The aim of IMspire 150 trial was to determine if combining checkpoint inhibitor with two targeted therapies would improve efficacy.
IMspire150 is a pivotal, placebo-controlled, international, multicenter, double-blinded, Phase III trial, in which 514 treatment-naive patients with Stage IIIc and Stage IV, BRAF V600–mutant malignant melanoma were enrolled. Patients were randomly assigned 1:1 to treatment with the doublet combination or the triplet therapy. Doublet therapy given to the control group of patients consisted of ZELBORAF® 960 mg orally twice daily plus COTELLIC® at 60 mg orally, on days 1 to 21 of a 28 day cycle. In the experimental or triplet therapy group, there was a 28-day run-in with ZELBORAF® plus COTELLIC® alone, dosed similar to the control group (cycle 1), following which patients received TECENTRIQ® 840 mg IV on Days 1 and 15 of each 28 day cycle starting cycle 2, in combination with ZELBORAF® at a lower dose of 720 mg orally twice daily and COTELLIC® 60 mg orally once daily. Treatment was continued until disease progression, or unacceptable toxicity. Both treatment groups were well balanced, median patient age was 54 years, 58% were male and 94% of patients had Stage IV disease. The Primary endpoint was investigator-assessed Progression Free Survival (PFS). Secondary end points included Objective Response Rates (ORR), Duration of Response (DOR), and Overall Survival (OS).
The combination of immunotherapy with targeted therapies was significantly superior to targeted therapies alone. At a median follow up of 18.9 months, the median PFS with the triplet combination was 15.1 months versus 10.6 months with the doublet therapy (HR=0.78; P=0.025). This represented a 22% reduction in the risk of disease progression. This benefit was observed across all subgroups including age, disease burden, LDH level, and extent of tumor involvement by organ site. Although Objective Response Rates were similar in both treatment groups, the median Duration of Response was 21.0 months with triplet combination versus 12.6 months for the doublet therapy. The OS data were not mature at the time of this analysis, but interim analysis however showed a median OS of 28.8 months with the triplet combination versus 25.1 months with doublet therapy. Both treatment groups had comparable toxicities. Among those patients receiving triplet combination, the most common toxicities were rash, fever, fatigue, nausea, pruritus, stomatitis, musculoskeletal pain, hepatotoxicity, edema, hypothyroidism, and photosensitivity.
It was concluded that in treatment-naive patients with advanced BRAF V600-mutant malignant melanoma, TECENTRIQ® in combination with ZELBORAF® and COTELLIC® significantly and clinically improved Progression Free Survival, when compared to placebo in combination with ZELBORAF® and COTELLIC®.
Evaluation of atezolizumab (A), cobimetinib (C), and vemurafenib (V) in previously untreated patients with BRAFV600 mutation-positive advanced melanoma: Primary results from the phase 3 IMspire150 trial. McArthur GA, Stroyakovskiy D, Gogas H, et al. Presented at: the 2020 AACR Annual Virtual Meeting I; April 27-28, 2020. Abstract CT012.