SUMMARY: The American Cancer Society estimates that for 2021, about 83,730 new cases of bladder cancer will be diagnosed in the US and about 17,200 patients will die of the disease. Bladder cancer is the fourth most common cancer in men, but is less common in women and the average age at the time of diagnosis is 73. With regards to racial predisposition, Caucasians are more likely to be diagnosed with bladder cancer than African Americans or Hispanic Americans.
Approximately 50% of all bladder cancers are non-invasive or in situ cancers. Patients with high-risk, Non-Muscle Invasive Bladder Cancer that has become unresponsive to BCG (Bacillus Calmette-Guerin) treatment, are often given the treatment option of radical cystectomy, which includes removing the entire urinary bladder and a prostatectomy for men or total hysterectomy in women. While highly curative, this surgical procedure carries substantial risk for morbidity and mortality, and can negatively impact patient’s quality of life. Further, a significant proportion of patients are medically ineligible for a radical cystectomy, and even if eligible, refuse surgery and opt for other less effective treatments, which could compromise outcomes.
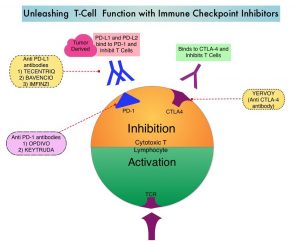
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. By doing so, it unleashes the tumor-specific effector T cells, and is thereby able to undo PD-1 pathway-mediated inhibition of the immune response. KEYTRUDA® is presently approved by the FDA for the treatment of patients with locally advanced or metastatic Urothelial carcinoma who are not eligible for Cisplatin-containing chemotherapy or for those with disease progression during or following platinum-containing chemotherapy, based on its durable antitumor activity in this patient group. Upregulation of the PD-1 pathway has been observed in BCG-resistant NMIBC, suggesting that KEYTRUDA® may be of benefit in this group of patients. The FDA in 2020, approved KEYTRUDA® for the treatment of patients with BCG-unresponsive, high-risk, Non-Muscle Invasive Bladder Cancer (NMIBC) with Carcinoma In Situ (CIS) with or without papillary tumors, who are ineligible for or have elected not to undergo cystectomy, based on the KEYNOTE-057 study.
KEYNOTE-057 study is an international, multicenter, single arm, open-label, Phase II trial that enrolled 101 eligible patients with high-risk NMIBC, who had BCG-unresponsive CIS with or without papillary tumors. BCG-unresponsive high-risk Non-Muscle Invasive Bladder Cancer was defined as persistent disease despite adequate BCG therapy, disease recurrence after an initial tumor-free state following adequate BCG therapy, or T1 disease following a single induction course of BCG. Eligible patients had received adequate BCG therapy and were unable/unwilling to undergo radical cystectomy. All patients had undergone TransUrethral Resection of Bladder Tumor (TURBT) to remove resectable disease. Patients with residual Carcinoma In Situ, not amenable to complete resection were permitted. Patients received KEYTRUDA® 200 mg IV every 3 weeks until unacceptable toxicity, persistent or recurrent high-risk NMIBC or progressive disease, or up to 24 months of therapy without disease progression. The efficacy analysis included 96 patients, as 5 patients did not meet the FDA BCG-unresponsive criteria.
The median age was 73 years and the median number of prior BCG instillations was 12. More than half of patients (56.9%) had a PD-L1 Combined Positive Score (CPS) of less than 10, and most patients in this analysis had refused prior cystectomy. The Primary end point was Complete Response Rate (CRR-absence of high-risk non-muscle invasive bladder cancer or progressive disease) as defined by negative results for cystoscopy with TURBT/biopsies as applicable, urine cytology, and CT Urography imaging, approximately 3 months after the first dose of KEYTRUDA®. Secondary end points included Duration of Response and Safety.
At a median follow up of 36.4 months, the Complete Response Rate at 3 months was 41% and the median Duration of Response was 16.2 months. Forty-six percent (46%) of responding patients experienced a Complete Response lasting 12 months or more. The Complete Response Rate was consistent with the primary analysis across protocol-prespecified subgroups, including PD-L1 expression status. The most frequent adverse reactions were fatigue, diarrhea, rash, pruritis, arthralgia, musculoskeletal pain, peripheral edema and hypothyroidism.
It was concluded that with extended follow up, KEYTRUDA® monotherapy continued to show durable and clinically meaningful antitumor activity in patients with high risk BCG-unresponsive, non-muscle invasive bladder cancer, who declined or were ineligible for radical cystectomy. The authors added that KEYTRUDA® should be considered a clinically active non-surgical treatment option in this difficult-to-treat population.
Pembrolizumab monotherapy for the treatment of high-risk non-muscle-invasive bladder cancer unresponsive to BCG (KEYNOTE-057): an open-label, single-arm, multicentre, phase 2 study. Balar AV, Kamat AM, Kulkarni GS, et al. The Lancet Oncology. 2021;22:919-930.