
The FDA on May 8, 2020, expanded the indication of LYNPARZA® to include its combination with Bevacizumab for first-line maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in Complete or Partial Response to first-line Platinum-based chemotherapy, and whose cancer is associated with Homologous Recombination Deficiency (HRD) positive status, defined by either a deleterious or suspected deleterious BRCA mutation, and/or genomic instability. LYNPARZA® is a product of AstraZeneca Pharmaceuticals, LP.
Author: RR
RETEVMO® (Selpercatinib)
The FDA on May 8, 2020, granted accelerated approval to RETEVMO® for the following indications:
1) Adult patients with metastatic RET fusion-positive Non-Small Cell Lung Cancer (NSCLC).
2) Adult and pediatric patients 12 years of age or older with advanced or metastatic RET-mutant Medullary Thyroid Cancer (MTC) who require systemic therapy.
3) Adult and pediatric patients 12 years of age or older with advanced or metastatic RET fusion-positive thyroid cancer who require systemic therapy, and who are Radioactive Iodine-refractory (if Radioactive Iodine is appropriate).
RETEVMO® is a product of Eli Lilly and Company.
FDA Approves TABRECTA® for Metastatic Non-Small Cell Lung Cancer
SUMMARY: The FDA on May 6, 2020, granted accelerated approval to TABRECTA® (Capmatinib) for adult patients with metastatic Non-Small Cell Lung Cancer (NSCLC), whose tumors have a mutation that leads to Mesenchymal-Epithelial Transition (MET) exon 14 skipping, as detected by an FDA-approved test. The FDA also approved the FoundationOne CDx assay (Foundation Medicine, Inc.) as a companion diagnostic for TABRECTA®.
MET is a widely expressed Receptor Tyrosine Kinase and plays a pivotal role in cell growth, proliferation and survival. The MET gene encodes for a protein known as the Hepatocyte Growth Factor (HGF) Receptor. Upon binding by Hepatocyte Growth Factor (HGF), the HGF Receptor is activated, with resulting activation of the downstream RAS/RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways, thereby serving different important biological functions. Alterations in the MET gene leading to abnormal MET signaling, has been identified in different types of cancers including thyroid, lung, breast, liver, colon, kidney, ovary and gastric carcinoma.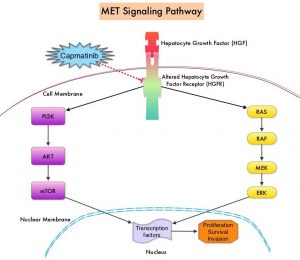
Two key MET alterations include MET exon 14 skipping mutations and MET amplification. MET exon 14 skipping mutations occur in approximately 5% of NSCLC patients with enrichment in sarcomatoid lung cancers (22%). MET exon 14 skipping mutation is a recognized oncogenic driver and is a molecular genetic abnormality indicating the presence of a splice site mutation that results in a loss of transcription of exon 14 of the MET gene. Most exon 14 mutations occur in never-smokers and is seen in both squamous and adenocarcinoma histology. Patients whose cancers have MET exon 14 skipping generally have very high response rates to MET inhibitors and molecular testing for MET exon 14 skipping should therefore be performed on all lung cancers, because this is a targetable alteration. MET amplification has been more commonly seen in smokers, and responses in patients with MET-amplified tumors might be more variable and dependent on level of amplification, with higher responses noted in tumors with more than 5-6 fold amplification. Tumors with MET exon 14 skipping mutations usually do not harbor activating mutations in EGFR, KRAS, or BRAF or concurrent ALK, ROS1 or RET translocations. However, it appears that cMET exon 14 skipping is not mutually exclusive with cMET amplification.
TABRECTA® (Capmatinib) is a highly potent and selective, reversible inhibitor of MET tyrosine kinase. The present FDA approval was based on the primary findings from the Phase II GEOMETRY mono-1 trial, which is a non-randomized, open-label, multi-cohort, Phase II study, conducted to evaluate the efficacy and safety of single-agent TABRECTA® in adult patients with EGFR wild-type, ALK-negative, metastatic NSCLC, whose tumors have a mutation that leads to MET exon 14 skipping (METex14), as detected by an RNA-based RT-PCR. This study enrolled 97 patients with metastatic NSCLC and confirmed MET exon 14 skipping mutations, 69 of whom were previously treated and, 28 of whom, were treatment naive. The patients received TABRECTA® at 400 mg orally twice daily until disease progression or unacceptable toxicity. The median patient age was 71 years and all NSCLC histologies including sarcomatoid/carcinosarcoma were included. Majority of the patients (75%) were white and 24% were Asian. Previous treatments included immunotherapy (28%) and chemotherapy (94%), and 23% of patients received 2 prior lines of therapy. The main efficacy outcome was Overall Response Rate (ORR) and additional efficacy outcomes included Duration of Response, Time to Response, Disease Control Rate, Progression Free Survival (PFS) and Safety. Thirteen patients (N=13) in this study had brain metastases at baseline.
Among the treatment-naïve patients group, the ORR was 68% with a median Duration of Response of 12.6 months and the percentage of patients with responses for 12 months or longer was 47%. The Disease Control Rate (Complete Response plus Partial Response plus Stable Disease) was 96.4%.
Among the previously treated patients, the ORR was 41%, with a median Duration of Response of 9.7 months and the percentage of patients with responses for 12 months or longer was 32%. The Disease Control Rate was 78.3%. Among those with brain metastases at baseline, 54% had an intracranial response with TABRECTA® with 31% showing complete resolution, 23% showing partial resolution, and the intracranial Disease Control Rate was 92%. The most common adverse events (occurring in at least 20% of patients) were peripheral edema, nausea, fatigue, vomiting, dyspnea, and decreased appetite. TABRECTA® can also cause Interstitial Lung Disease, hepatotoxicity and photosensitivity.
It was concluded that TABRECTA® is a new treatment option for patients with MET exon 14 skipping- mutated advanced NSCLC, regardless of the line of therapy, with deep and durable responses, manageable toxicity profile, and is the first and only FDA approved treatment for this patient group.
Capmatinib (INC280) in METex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. Wolf J, Seto T, Han J, et al. J Clin Oncol. 2019;37(suppl; abstr 9004).
Proton Based Chemoradiotherapy Significantly Decreases Toxicities without Compromising Efficacy
SUMMARY: Radiation Therapy involves the use of X-Rays, Gamma rays and charged particles for cancer treatment. External Beam Radiation Therapy (EBRT) is most often delivered using a linear accelerator in the form of Photon beams (either X-rays or Gamma rays). Photons have no mass and are packets of energy of an electromagnetic wave. Electrons and Protons are charged particles and Electrons are considered light particles whereas Protons are considered heavy particles. Electron beams are used to irradiate skin and superficial tumors, as they are unable to penetrate deep into the tissues. The different types of External Beam Radiation Treatments include 3-Dimensional Conformal Radiation Therapy (3D-CRT) meant to deliver radiation to very precisely shaped target areas, IMRT or Intensity Modulated Radiation Therapy which allows different areas of a tumor or nearby tissues to receive different doses of radiation, Image Guided Radiation Therapy (IGRT) which allows reduction in the planned volume of tissue to be treated, as changes in a tumor size are noted during treatment, Stereotactic RadioSurgery (SRS) which can deliver one or more high doses of radiation to a small tumor and Stereotactic Body Radiation Therapy (SBRT) or CYBERKNIFE® which is similar to SRS but also takes the normal motion of the body into account while treating malignancies involving the lung and liver.
Proton beams unlike Photons, enter the skin and travel through the tissues and deposit much of their energy at the end of their path (known as the Bragg peak), and deposit less energy along the way. This is unlike Photons which deposit energy all along the path through the tissues and the deposited dose decreases with increasing depth. As a result, with Proton beam therapy, normal tissues are exposed to less radiation compared with Photons. Despite this advantage, tissue heterogeneity such as organ motion, tumor volume changes during treatment can have a significant negative impact on target coverage for Proton beam therapy and can result in damage to the surrounding tissues and potential complications. It is well established that there is significant benefit for Proton beam therapy in certain pediatric malignancies.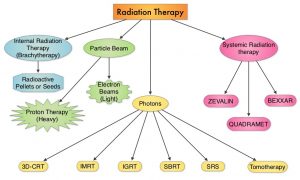
Curative treatment with concurrent chemoradiotherapy is the standard of care for many nonmetastatic, locally advanced cancers. This treatment modality however is associated with substantial morbidity. Proton therapy as component of concurrent chemoradiotherapy might be able to reduce treatment related toxicity and achieve comparable cancer control outcomes, compared with conventional Photon radiotherapy, by reducing the radiation dose to normal tissues. There are however limited data comparing results of Proton chemoradiotherapy with Photon chemoradiotherapy, and Proton therapy remains unproven in this treatment setting. The objective of this study was to assess whether Proton therapy in the setting of concurrent chemoradiotherapy is associated with fewer hospitalizations or other adverse events and similar Disease-free and Overall Survival, compared with concurrent Photon chemoradiotherapy.
In this large single-institution, nonrandomized, comparative effectiveness, retrospective analysis, 1483 adult patients with nonmetastatic, locally advanced cancer, treated with concurrent chemoradiotherapy with curative intent were included. Three hundred ninety-one patients (N=391) received Proton therapy and 1092 patients received Photon therapy. Common tumor sites included head and neck, lung, brain, esophagus/gastric, rectum, and pancreas. The median patient age was 62 years, but patients treated with Protons were significantly older with a median age of 66 years versus 61 years, had less favorable Charlson-Deyo comorbidity scores and had lower integral radiation dose to tissues outside the target. Ninety three percent (93%) of patients in the Photon therapy group were treated with Intensity-Modulated Radiotherapy (IMRT). Baseline ECOG Performance Status was similar between the two treatment cohorts. The Primary end point was 90-day adverse events associated with unplanned hospitalizations (CTCAE version 4 – Grade 3 or more). Secondary end points included ECOG performance status decline during treatment, 90-day adverse events of at least Grade 2 that limit instrumental activities of daily living, and Disease-Free and Overall Survival. The data on adverse events and survival were gathered prospectively.
It was noted that Proton chemoradiotherapy was associated with a significantly lower relative risk of 90-day adverse events of at least Grade 3 (P=0.002), significantly lower relative risk of 90-day adverse events of at least Grade 2 (P=0.006), and decline in Performance Status during treatment (P<0.001). Proton chemoradiotherapy was associated with a two-thirds reduction in adverse events associated with unplanned hospitalizations. At a median follow up of 3.7 years for the Proton cohort and 4.2 years for the Photon cohort, there was no difference in Disease-Free or Overall Survival.
It was concluded from this analysis that in adults with locally advanced cancer, Proton chemoradiotherapy was associated with significantly reduced acute adverse events that caused unplanned hospitalizations, with similar Disease-Free and Overall Survival, compared to Photon therapy.
Comparative Effectiveness of Proton vs Photon Therapy as Part of Concurrent Chemoradiotherapy for Locally Advanced Cancer. Baumann BC, Mitra N, Harton JG, et al. Jama Oncol. 2020;6:237-246.
FDA Approves ZEJULA® for Newly Diagnosed Patients with Advanced Ovarian Cancer
SUMMARY: The FDA on April 29, 2020 approved ZEJULA® (Niraparib) for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a complete or partial response to first-line platinum-based chemotherapy. It is estimated that in the United States, approximately 21,750 women will be diagnosed with ovarian cancer in 2020 and 13,940 women will die of the disease. Ovarian cancer ranks fifth in cancer deaths among women, and accounts for more deaths than any other cancer of the female reproductive system. Approximately 75% of the ovarian cancer patients are diagnosed with advanced disease. Patients with newly diagnosed advanced ovarian cancer are often treated with platinum based chemotherapy following primary surgical cytoreduction. Approximately 70% of these patients will relapse within the subsequent 3 years and are incurable, with a 5 year Overall Survival rate of about 20-30%.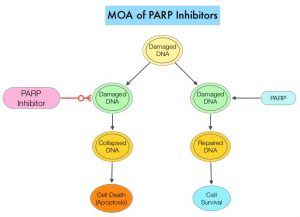
DNA damage is a common occurrence in daily life by UV light, ionizing radiation, replication errors, chemical agents, etc. This can result in single and double strand breaks in the DNA structure which must be repaired for cell survival. The two vital pathways for DNA repair in a normal cell are BRCA1/BRCA2 and PARP. The PARP (Poly ADP Ribose Polymerase) family of enzymes, include PARP1 and PARP2. In the context of DNA repair, BRCA1 and BRCA2 genes recognize and repair double strand DNA breaks via Homologous Recombination (HR) pathway. Homologous Recombination is a type of genetic recombination, and is a DNA repair pathway utilized by cells to accurately repair DNA double-stranded breaks during the S and G2 phases of the cell cycle, and thereby maintain genomic integrity. Homologous Recombination Deficiency (HRD) is noted following mutation of genes involved in HR repair pathway. At least 15 genes are involved in the Homologous Recombination Repair (HRR) pathway including BRCA1 and BRCA2 genes. The BRCA1 gene is located on the long (q) arm of chromosome 17 whereas BRCA2 is located on the long arm of chromosome 13. BRCA1 and BRCA2 are tumor suppressor genes and functional BRCA proteins repair damaged DNA, and play an important role in maintaining cellular genetic integrity. They regulate cell growth and prevent abnormal cell division and development of malignancy. Mutations in BRCA1 and BRCA2 account for about 20-25% of hereditary breast cancers and about 5-10% of all breast cancers. They also account for 15% of ovarian cancers, in addition to other cancers such as Colon and Prostate. BRCA mutations can either be inherited (Germline) and present in all individual cells or can be acquired and occur exclusively in the tumor cells (Somatic). Somatic mutations account for a significant portion of overall BRCA1 and BRCA2 aberrations. Loss of BRCA function due to frequent somatic aberrations likely deregulates HR pathway, and other pathways then come in to play, which are less precise and error prone, resulting in the accumulation of additional mutations and chromosomal instability in the cell, with subsequent malignant transformation. HRD therefore indicates an important loss of DNA repair function. Hereditary Epithelial Ovarian Cancer was thought to be caused almost exclusively by mutations in BRCA1 and BRCA2. It however is now well known that about 50% of the high grade serous ovarian cancers have aberrations in HR repair pathway. Deregulated HR pathway increases sensitivity to platinum drugs. Majority of the women with germline BRCA mutations (gBRCA) are positive for HR deficiency.
PARP is a related enzymatic pathway that repairs single strand breaks in DNA. In a BRCA mutant, the cancer cell relies solely on PARP pathway for DNA repair. In the presence of a PARP inhibitor, there is synthetic lethality because loss of both genes, leading to cell death. Thus PARP inhibitors are only harmful to cancer cells. ZEJULA® is a highly selective PARP 1/2 inhibitor, that causes cumulative DNA damage and cell death by inhibiting PARP. Previously published phase III study among patients with platinum-sensitive, recurrent ovarian cancer (NEJM 2016;375:2154-2164) concluded that Niraparib significantly prolonged Progression Free Survival (PFS) compared to placebo, and this benefit was achieved regardless of the presence or absence of germline BRCA mutations or HRD status.
PRIMA trial is a randomized, double-blind, placebo-controlled, international Phase III trial conducted to test the efficacy and safety of ZEJULA® maintenance therapy after a response to platinum-based chemotherapy, in patients with newly diagnosed advanced ovarian cancer at high risk for relapse. It should be noted that at the time PRIMA trial was designed, AVASTIN® (Bevacizumab) was not approved for first-line treatment in all participating countries. A total of 733 patients with newly diagnosed, high risk, advanced ovarian cancer were randomly assigned in a 2:1 ratio to receive ZEJULA® (N=487) or placebo (N=246) once daily in 28-day cycles for 36 months or until disease progression, after a response to platinum-based chemotherapy regimen. Patients received a dose of 200-300mg once daily, based on body weight and platelet count. Enrolled patients were at high risk for progressive disease with 23.1% having Stage III ovarian cancer with residual disease after primary debulking surgery, 66.7% had received neoadjuvant chemotherapy, 35% had Stage IV ovarian cancer, and 30.5% had a Partial Response to first-line platinum-based chemotherapy. Tumor samples were tested for HRD status and HRD was defined by either presence of tumor BRCA mutation or Genomic Instability Score (GIS) of 42 or more. Of the randomized patients, 50.9% had tumors with HRD, 30.4% had BRCA mutations and 20.5% were BRCA wild type. The treatment groups were well balanced. The Primary endpoint was Progression Free Survival (PFS) in patients who had tumors with HRD, and then in the overall population, as determined on hierarchical testing. Secondary end points included Overall Survival, time until the first subsequent therapy, PFS 2, defined as time from randomization to progression while the patient was receiving a subsequent anticancer therapy and Patient-Reported Outcomes. The median duration of follow-up at the time of the data cutoff was 13.8 months.
There was a statistically significant improvement in PFS for patients randomized to ZEJULA® compared with placebo in the HRD group, as well as the overall population. The median PFS in the HRD group was 21.9 months for patients receiving ZEJULA® compared with 10.4 months for those receiving placebo (HR=0.43; P<0.001). The median PFS in the overall population was 13.8 months for patients receiving ZEJULA® compared with 8.2 months for those receiving placebo (HR=0.62; P<0.001). At the 24-month interim analysis, the rate of Overall Survival was 84% in the ZEJULA® group and 77% in the placebo group (HR=0.70). The most common adverse reactions in patients receiving ZEJULA® were cytopenias, fatigue, AST/ALT elevation, hypertension, low grade nausea and decreased appetite.
It was concluded that among patients with newly diagnosed advanced ovarian cancer who had responded to platinum-based chemotherapy, ZEJULA® significantly prolonged Progression Free Survival, compared to those who received placebo, regardless of the presence or absence of Homologous Recombination Deficiency.
Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. González-Martín A, Pothuri B, Vergote I, et al. for the PRIMA/ENGOT-OV26/GOG-3012 Investigators. N Engl J Med 2019; 381:2391-2402
FDA Approves Subcutaneous DARZALEX® in Multiple Myeloma
SUMMARY: The FDA on May 1, 2020 approved DARZALEX® (Daratumumab) and Hyaluronidase-fihj (DARZALEX FASPRO®), for adult patients with newly diagnosed or Relapsed/Refractory multiple myeloma. This new product allows for subcutaneous dosing of DARZALEX®.
DARZALEX FASPRO® is now approved for these previously approved indications for IV DARZALEX®
1) In combination with VELCADE® (Bortezomib), Melphalan and Prednisone in newly diagnosed patients who are ineligible for Autologous Stem Cell Transplant (ASCT)
2) In combination with REVLIMID® (Lenalidomide) and Dexamethasone in newly diagnosed patients, who are ineligible for ASCT and in patients with Relapsed or Refractory multiple myeloma who have received at least one prior therapy
3) In combination with VELCADE® and Dexamethasone in patients who have received at least one prior therapy
4) As monotherapy, in patients who have received at least three prior lines of therapy including a Proteasome Inhibitor (PI) and an Immunomodulatory agent or who are double-refractory to a PI and an immunomodulatory agent.
DARZALEX® is a human IgG1 antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Dependent Cytotoxicity (CDC) and direct Apoptosis. Additionally, DARZALEX® may play a role in immunomodulation, by depleting CD38-positive regulator immune suppressor cells, and thereby expanding T cells, in patients responding to therapy.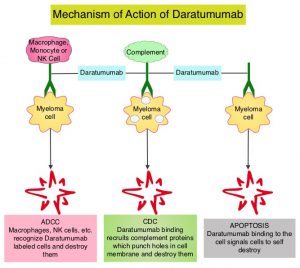
This FDA approval is based on COLUMBA Trial, which is a randomized, open-label, multicenter Phase III study, which included 522 patients with multiple myeloma, who had received at least three prior lines of therapy including a Proteasome Inhibitor (PI) and an immunomodulatory drug (IMiD), or whose disease was refractory to both a PI and an IMiD. Patients were randomly assigned to receive a fixed dose of subcutaneously (SC) administered formulation of DARZALEX® 1800 mg weekly for cycles 1-2, every two weeks for cycles 3-6 and every four weeks for cycle 7 and thereafter (N=263), with the subcutaneous preparation given over 3-5 minutes at alternating left and right abdominal sites. In the intravenous group, patients received DARZALEX® 16 mg/kg IV weekly for cycles 1-2, every two weeks for cycles 3-6 and every four weeks for cycle 7 and thereafter (N=259). Each cycle was 28 days. Treatment in both patient groups was continued until disease progression or unacceptable toxicity. The median age was 67 years and the median number of prior therapies was four in each treatment group. Patient characteristics were similar between the two arms except that more patients in the subcutaneous arm had high-risk cytogenetics (26%) compared with the intravenous group (17%). The median duration of treatment was approximately 5 months, with a median of 6 completed cycles of treatment. The median duration of infusion was consistently 5 minutes at each visit in the subcutaneous group. However, in the IV arm, the first infusion lasted 7 hours, the second infusion was 4.3 hours, and subsequent infusions lasted a median of 3.4 hours. The study co-Primary endpoints were Overall Response Rate (ORR) and pharmacokinetic endpoint of the maximum C-trough on cycle 3, day 1 pre-dose.
At a median follow up of 7.5 months, the ORR was 41% for the subcutaneous administered formulation of DARZALEX® compared to 37% for IV DARZALEX® (P<0.0001). The ORR was similar across all clinically relevant subgroups, including body weight. The ratio of geometric means of C-trough for the SC administered formulation of DARZALEX® over IV DARZALEX® was 108%. The Progression Free Survival was comparable between the SC administered formulation of DARZALEX and the current IV formulation of DARZALEX (HR=0.99; P<0.9258). A lower rate of infusion-related reactions was observed in the group that received the SC DARZALEX® compared to IV DARZALEX® (13% vs. 35%, respectively).
It was concluded that the subcutaneous formulation of DARZALEX® resulted in non-inferior pharmacokinetics and efficacy compared to the current IV formulation, and also importantly offers the potential for a fixed-dose administration, shorter administration times and a lower rate of infusion-related reactions with improved safety profile, in patients with Relapsed or Refractory multiple myeloma.
Subcutaneous versus intravenous daratumumab in patients with relapsed or refractory multiple myeloma (COLUMBA): a multicentre, open-label, non-inferiority, randomised, phase 3 trial. Mateos M-V, Nahi H, Legiec W, et al. The Lancet Haematology. Published: March 23, 2020. DOI: https://doi.org/10.1016/S2352-3026(20)30070-3.
TABRECTA® (Capmatinib)
The FDA on May 6,2020 granted accelerated approval to TABRECTA® for adult patients with metastatic Non-Small Cell Lung Cancer (NSCLC) whose tumors have a mutation that leads to Mesenchymal-Epithelial Transition (MET) exon 14 skipping, as detected by an FDA-approved test. TABRECTA® is a product of Novartis.
DARZALEX FASPRO® (Daratumumab and Hyaluronidase-fihj)
The FDA on May 1, 2020 approved DARZALEX FASPRO® for adult patients with newly diagnosed or Relapsed/Refractory Multiple Myeloma. This new product allows for subcutaneous dosing of Daratumumab. DARZALEX FASPRO® is a product of Janssen Biotech, Inc.