SUMMARY: Colorectal Cancer (CRC) is the third most common cancer diagnosed in both men and women in the United States. The American Cancer Society estimates that approximately 147,950 new cases of CRC will be diagnosed in the United States in 2020 and about 53,200 patients are expected to die of the disease. The lifetime risk of developing CRC is about 1 in 23. The majority of CRC cases (about 75 %) are sporadic whereas the remaining 25 % of the patients have family histories of the disease. Only 5-6 % of patients with CRC with a family history background are due to inherited mutations in major CRC genes, while the rest are the result of accumulation of both genetic mutations and epigenetic modifications of several genes.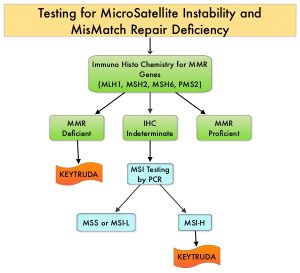
The DNA MisMatchRepair (MMR) system is responsible for molecular surveillance and works as an editing tool that identifies errors within the microsatellite regions of DNA and removes them. Defective MMR system leads to MSI (Micro Satellite Instability) and hypermutation, triggering an enhanced antitumor immune response. MSI is therefore a hallmark of defective/deficient DNA MisMatchRepair (dMMR) system and occurs in 15% of all colorectal cancers. Defective MMR can be a sporadic or heritable event. Approximately 65% of the MSI colon tumors are sporadic and when sporadic, the DNA MMR gene is MLH1. Defective MMR can manifest as a germline mutation occurring in MMR genes including MLH1, MSH2, MSH6 and PMS2. This produces Lynch Syndrome often called Hereditary Nonpolyposis Colorectal Carcinoma – HNPCC, an Autosomal Dominant disorder that is often associated with a high risk for Colorectal and Endometrial carcinoma, as well as several other malignancies including Ovary, Stomach, Small bowel, Hepatobiliary tract, Brain and Skin. MSI is a hallmark of Lynch Syndrome-associated cancers. MSI tumors tend to have better outcomes and this has been attributed to the abundance of tumor infiltrating lymphocytes in these tumors from increase immunogenicity. These tumors therefore are susceptible to blockade with immune checkpoint inhibitors. MSI testing is performed using a PCR based assay and MSI-High refers to instability at 2 or more of the 5 mononucleotide repeat markers and MSI-Low refers to instability at 1 of the 5 markers. Patients are considered Micro Satellite Stable (MSS) if no instability occurs. MSI-L and MSS are grouped together because MSI-L tumors are uncommon and behave similar to MSS tumors. Tumors considered MSI-H have deficiency of one or more of the DNA MMR genes. MMR gene deficiency can be detected by ImmunoHistoChemistry (IHC). NCCN Guidelines recommend MMR or MSI testing for all patients with a history of Colon or Rectal cancer. Unlike Colorectal and Endometrial cancer, where MSI-H/dMMR testing is routinely undertaken, the characterization of Lynch Syndrome across heterogeneous MSI-H/dMMR tumors is unknown.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells. The FDA in 2017 granted accelerated approval to KEYTRUDA® for patients with advanced MSI-High or dMMR solid tumors, that have progressed following prior treatment, and who have no satisfactory alternative treatment options. This has led to routine MSI-H/dMMR testing in advanced solid tumors.
KEYNOTE-177 is an International, multicenter, randomized open-label, Phase III trial conducted, to evaluate the efficacy and safety of KEYTRUDA® versus Standard-of-Care (SOC) chemotherapy, as first-line therapy for dMMR or MSI-H metastatic ColoRectal Cancer (mCRC). In this study, a total of 307 patients with MSI-H/dMMR mCRC as determined locally, and with ECOG PS of 0 or 1 were randomly assigned 1:1 to first-line treatment with KEYTRUDA® 200 mg IV every 3 weeks for up to 2 years (N=153) or investigator’s choice of mFOLFOX-6 or FOLFIRI every 2 weeks, with or without Bevacizumab or Cetuximab (N=154). Chemotherapy regimens were chosen prior to randomization. Treatment was continued until disease progression, unacceptable toxicity or completion of 35 cycles (for KEYTRUDA® only). The median patient age was 63 years and both treatment groups were well balanced. The co-Primary endpoints of the study were Progression Free Survival (PFS) and Overall Survival (OS). Key Secondary endpoints included Overall Response Rate (ORR) and Safety. Patients with confirmed disease progression on chemotherapy were given the option to crossover, to receive treatment with KEYTRUDA®. The median follow up was 28 months.
It was noted that KEYTRUDA® was superior to chemotherapy with a median PFS of 16.5 months versus 8.2 months for chemotherapy (HR=0.60; P=0.0002). The 12 and 24-months PFS rates were 55.3% and 48.3% with KEYTRUDA® versus 37.3% and 18.6% with chemotherapy, respectively. The confirmed ORR was 43.8% with KEYTRUDA® versus 33.1% with chemotherapy and the median Duration of Response was not reached in the KEYTRUDA® group and was 10.6 months in the chemotherapy group. Following disease progression, 36% of patients assigned to the chemotherapy group crossed over to the KEYTRUDA® group. This study is being continued to evaluate OS. Grade 3-5 treatment related Adverse Event rates were 22% in the KEYTRUDA® arm and 66% in the chemotherapy group.
The authors concluded that when compared to chemotherapy, first-line therapy with KEYTRUDA® provided a clinically meaningful and statistically significant improvement in Progression Free Survival, among patients with MSI-H/dMMR metastatic colorectal cancer, with fewer treatment-related Adverse Events. The authors added that KEYTRUDA® should be the new standard of care for this patient group.
Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE-177 study. Andre T, Shiu K-K, Kim TW, et al. J Clin Oncol 38: 2020 (suppl; abstr LBA4)