
The FDA on July 31, 2020 granted accelerated approval to MONJUVI®, a CD19-directed cytolytic antibody, indicated in combination with REVLIMID® (Lenalidomide), for adult patients with Relapsed or Refractory Diffuse Large B-cell Lymphoma (DLBCL) Not Otherwise Specified (NOS), including DLBCL arising from Low grade lymphoma, and who are not eligible for Autologous Stem Cell Transplant. MONJUVI® is a product of MorphoSys US Inc.
Author: RR
First Line CYRAMZA® Plus ERLOTINIB® for EGFR-Mutated Non Small Cell Lung Cancer
SUMMARY: The FDA on May 29, 2020 approved CYRAMZA® (Ramucirumab) in combination with TARCEVA® (Erlotinib) for first line treatment of metastatic Non Small Cell Lung Cancer (NSCLC) with Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) mutations.
Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R substitution mutation in Exon 21. EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to the most common, acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients.
TAGRISSO® (Osimertinib) is a highly selective third-generation Epidermal Growth Factor Receptor (EGFR) TKI presently approved by the FDA, for the first-line treatment of patients with metastatic NSCLC, whose tumors have Exon 19 deletions or Exon 21 L858R mutations, as well as treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, whose disease has progressed on or after EGFR-TKI therapy. Previously published data from the Phase III FLAURA study showed that first-line treatment with TAGRISSO® was superior to first-line treatment with other first and second generation TKI’s, in patients with EGFR-mutated NSCLC, with improved median Overall Survival. Patients with the exon 21 L858R substitution mutation however are less sensitive to TKIs and their PFS therefore has been lower, than those with exon 19 deletions. In the FLAURA study the PFS in the exon 21 mutation group was 14.4 months compared with 21.4 months for patients with an exon 19 deletion, when treated with first line TAGRISSO®. Furthermore, widespread use of TAGRISSO® has led to acquired resistance. Evolving data has shown limited responses to immune checkpoint inhibitors after disease progression on TKIs. There is therefore a critical need to develop new TKI-based therapies, and novel treatment approaches, combining TKI’s with other targeted therapies. RELAY trial was designed to address this unmet need.
CYRAMZA® is a recombinant human monoclonal IgG1 antibody that binds to the human Vascular Endothelial Growth Factor Receptor- 2 (VEGFR-2), preventing the interaction of VEGFR-2 with its ligands. CYRAMZA® was previously approved for use in combination with TAXOTERE® (Docetaxel) for the treatment of patients with metastatic NSCLC who progressed while on or following treatment with platinum-based chemotherapy. Several preclinical studies have shown that dual blockade of the EGFR and VEGF pathways in EGFR-mutated metastatic NSCLC is synergistic, with higher antitumor activity, when compared with inhibition of the EGFR pathway alone. This has been attributed to upregulation of VEGF in the tumor microenvironment, when tumor cells harbor EGFR mutations.
RELAY trial is global, multicenter, randomized, double-blind, placebo-controlled, Phase III study in which 449 patients with previously untreated metastatic NSCLC, who harbored either an EGFR exon 19 deletion or exon 21 L858R substitution mutation, were enrolled. Patients were randomized 1:1 to receive TARCEVA® 150 mg orally daily in combination with either CYRAMZA® 10 mg/kg IV (N=224) or placebo (N=225), every 2 weeks. Treatment was continued until disease progression or unacceptable toxicity. Both treatment groups were well balanced. Patients had an ECOG performance status of 0-1, median age was 64 years, and about 60% of patients were never smokers. Those with a known EGFR T790M mutation, received prior treatment with an EGFR TKI or chemotherapy, or had brain metastases, were ineligible for study enrollment. Patients were stratified by sex, EGFR mutation type, and EGFR testing methodology. The Primary end point was Progression Free Survival (PFS). Secondary end points included Overall Survival (OS), Overall Response Rate (ORR) and Duration of Response (DOR).
At a median follow up of 20.7 months, the median PFS was 19.4 months in the CYRAMZA® plus TARCEVA® group compared with 12.4 months in the placebo plus TARCEVA® group (HR= 0.59; P<0.0001). This PFS benefit was observed across several patient subgroups, and was consistent across Exon 19 and Exon 21 subgroups. Unlike in the FLAURA trial, in the RELAY trial, the PFS in patients with exon 21 mutation was comparable to patients with exon 19 deletions. The PFS for these patients with exon 21 mutation was 19.4 months. Further, the addition of CYRAMZA® to TARCEVA® did not increase the incidence of EGFR T790M mutation. The ORR was 76% in the CYRAMZA® plus TARCEVA® group and 75% in the placebo plus TARCEVA® group, with median DoR of 18.0 months and 11.1 months, respectively. At the time of the final analysis of PFS, the OS data were not mature. The most common adverse reactions in the CYRAMZA® plus TARCEVA® group were infections, hypertension, stomatitis, proteinuria, alopecia, epistaxis, and peripheral edema. The most common laboratory abnormalities were increased ALT and AST as well as cytopenias.
It was concluded that CYRAMZA® plus TARCEVA® demonstrated superior PFS, compared with placebo plus TARCEVA®, in treatment naïve patients with EGFR-mutated metastatic NSCLC. Inhibiting the VEGFR and EGFR pathways together, is an important milestone in the treatment of EGFR-mutated NSCLC, with outcomes comparable to that with third generation TKIs, and furthermore, providing these patients an additional treatment option with TAGRISSO® upon progression.
Ramucirumab plus Erlotinib in Patients with Untreated, EGFR-mutated, Advanced Non-Small-Cell Lung Cancer (RELAY): A Randomised, Double-blind, Placebo-Controlled, Phase 3 trial. Nakagawa K, Garon EB, Seto T, et al. Lancet Oncol. 2019;20:1655-1669.
FDA Approves MONJUVI® for Diffuse Large B-Cell Lymphoma
SUMMARY: The FDA on July 31, 2020, granted accelerated approval to MONJUVI® (Tafasitamab-cxix), a CD19-directed cytolytic antibody, in combination with REVLIMID® (Lenalidomide), for adult patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma (DLBCL) Not Otherwise Specified, including DLBCL arising from low grade lymphoma, and who are not eligible for Autologous Stem Cell Transplant.
The American Cancer Society estimates that in 2020, about 77,240 people will be diagnosed with Non Hodgkin Lymphoma (NHL) in the United States and about 19,940 individuals will die of this disease. Diffuse Large B-Cell Lymphoma (DLBCL) is the most common of the aggressive Non-Hodgkin lymphoma’s in the United States, and the incidence has steadily increased 3-4% each year. More than half of patients are 65 or older at the time of diagnosis and the incidence is likely to increase with the aging of the American population. The etiology of Diffuse Large B-Cell Lymphoma is unknown. Contributing risk factors include immunosuppression (AIDS, transplantation setting, autoimmune diseases), UltraViolet radiation, pesticides, hair dyes, and diet. DLBCL is a neoplasm of large B cells and the most common chromosome abnormality involves alterations of the BCL-6 gene at the 3q27 locus, which is critical for germinal center formation. Two major molecular subtypes of DLBCL arising from different genetic mechanisms have been identified, using Gene Expression Profiling: Germinal Center B-cell-like (GCB) and Activated B-Cell-like (ABC). Patients in the GCB subgroup have a higher five year survival rate, independent of clinical IPI (International Prognostic Index) risk score, whereas patients in the ABC subgroup have a significantly worse outcome. Regardless, R-CHOP regimen (RITUXAN®-Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone), given every 21 days, for 6 cycles, delivered with curative intent, is the current standard of care for patients of all ages, with newly diagnosed DLBCL, regardless of molecular subtype. Approximately 30-40% of patients experience disease progression or relapse, during the first 2 years and attempts to improve on R-CHOP regimen have not been successful. Maintenance treatment strategy following R-CHOP, to better control the disease, delay disease progression and improve long term survival, have included Autologous Stem Cell Transplantation, maintenance treatment with agents such as oral protein kinase inhibitor Enzastaurin and Everolimus. Outcomes for transplant-ineligible patients with Relapsed/Refractory DLBCL patients remain poor.
REVLIMID® (Lenalidomide) is an oral immunomodulatory agent with activity in lymphoid malignancies, primarily through immune modulation (repair T-cell immune synapse dysfunction and Natural Killer cell/T-cell effector augmentation). It additionally has antiproliferative effects. REVLIMID® was shown to have significant activity in relapsed DLBCL when given alone or along with RITUXAN®. MONJUVI® is an investigational humanized Fc-engineered monoclonal antibody directed against CD19. MONJUVI® incorporates an XmAb(R) engineered Fc domain, which is intended to lead to a significant potentiation of Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) and Antibody-Dependent Cellular Phagocytosis (ADCP), thus improving tumor cell kill. Preclinical data suggested that MONJUVI® might act synergistically with REVLIMID®.
L-MIND is an ongoing , multicenter, single arm, open-label, Phase II study, investigating the combination of MONJUVI® and REVLIMID® in patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma (R/R DLBCL), after up to two prior lines of therapy, including an anti-CD20 targeting therapy (such as Rituximab), who are not eligible for high-dose chemotherapy and subsequent Autologous Stem Cell Transplantation. This study enrolled 81 patients and patients received 28-day cycles of MONJUVI® 12 mg/kg IV once weekly during Cycles 1-3 with a loading dose on Cycle 1 Day 4, then every 2 weeks during Cycles 4-12, along with REVLIMID® 25 mg orally daily on Days 1-21 of Cycles 1-12. After Cycle 12, progression-free patients received MONJUVI® every 2 weeks until disease progression. Eighty patients (N=80) received at least one dose of both MONJUVI® and REVLIMID®. The Primary endpoint was Objective Response Rate (ORR). Secondary endpoints included Duration of Response (DoR), Progression-Free Survival (PFS) and Overall Survival (OS).
In this long-term analysis after a minimum of two years follow-up, outcomes from the L-MIND study were consistent with the primary analysis. Assessment by an Independent Review Committee at data cut-off showed an ORR of 58.8% and a Complete Response (CR) rate of 41.3%. Median Duration of Response was 34.6 months. The median OS was 31.6 months and median PFS was 16.2 months. The safety profile was consistent with that observed in previously reported studies of MONJUVI® in combination with REVLIMID®. The most common Grade 3 or worse Adverse Events were cytopenias and febrile neutropenia.
To determine the the contribution of MONJUVI® in the combination with REVLIMID® and to prove its synergistic effect, an observational retrospective study was conducted (Re-MIND) to compare real-world response data of patients with Relapsed or Refractory DLBCL who received REVLIMID® monotherapy with the efficacy outcomes of the MONJUVI®- REVLIMID® combination, as investigated in the L-MIND trial. In this study, efficacy data was collected from 490 R/R DLBCL patients in the US and EU. Qualification criteria for matching patients of both studies were pre-specified. As a result, 76 eligible Re-MIND patients were identified and matched 1:1 to 76 of 80 L-MIND patients based on important baseline characteristics. Objective response rates (ORR) were validated based on this subset of 76 patients in Re-MIND and L-MIND, respectively. The Primary endpoint of Re-MIND was met and shows a statistically significant superior best ORR of the MONJUVI®/ REVLIMID® combination compared to REVLIMID® monotherapy. Further, there was a significant difference in OS as well as CR rates, favoring the L-MIND cohort over the observational cohort.
It was concluded that MONJUVI® in combination with REVLIMID® resulted high Complete Response rates, as well Durable Responses and improved survival, in a significant proportion of patients with relapsed or refractory Diffuse Large B-Cell Lymphoma, ineligible for Autologous Stem Cell Transplantation, and might represent a new therapeutic option in this clinical setting.
Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Salles G, Duell J, Barca EG, et al. Lancet Oncol. 2020 Jul;21:978-988.
FDA Approves Oral INQOVI® for Myelodysplastic Syndromes
SUMMARY: The FDA on July 7, 2020, approved INQOVI®, an oral combination of Decitabine and Cedazuridine, for adult patients with MyeloDysplastic Syndromes (MDS), including previously treated and untreated de novo and secondary MDS with the following FAB subtypes – Refractory Anemia, Refractory Anemia with Ringed Sideroblasts, Refractory Anemia with Excess Blasts, Chronic MyeloMonocytic Leukemia (CMML), and Intermediate-1, Intermediate-2, and high-risk International Prognostic Scoring System (IPSS) groups.
It is estimated that in the US approximately 13,000 people are diagnosed with MyeloDysplastic Syndromes (MDS) each year. The prevalence has been estimated to be from 60,000 to 170,000 in the US. MyeloDysplastic Syndromes are a heterogenous group of stem cell disorders characterized by marrow failure resulting in cytopenias with associated cytogenetic abnormalities, and abnormal cellular maturation with morphologic changes in clonal cells. Majority of the individuals diagnosed with MDS are 65 years or older and die as a result of infection and/or bleeding, consequent to bone marrow failure. About a third of patients with MDS develop Acute Myeloid Leukemia (AML). CMML (Chronic MyeloMonocytic Leukemia) is a clonal hematopoietic malignancy characterized by accumulation of abnormal monocytes in the bone marrow and in blood. The incidence of CMML in the US is approximately 1,100 new cases per year. About 15-30% of patients with CMML develop AML. Patients with higher risk MDS and CMML are often treated with hypomethylating agents such as Decitabine (DACOGEN®) and Azacitidine (VIDAZA®). These agents are administered by IV infusion, or by large-volume subcutaneous injections.
INQOVI® is an orally-administered, unique fixed-dose combination of the DNA hypomethylating agent and DNA MethylTransferase (DNMT) inhibitor Decitabine, the active ingredient in Dacogen®, and the novel Cytidine deaminase inhibitor, Cedazuridine (35 mg Decitabine and 100 mg Cedazuridine). INQOVI® was designed to deliver Decitabine by oral administration. Cedazuridine prevents the degradation of Decitabine in the gut and liver by inhibiting Cytidine deaminase and the combination thereby permits the efficient delivery of Decitabine orally, at exposures that are equivalent to the approved intravenous form of Decitabine administered over 5 days.
The present FDA approval was based on data from two open-label, randomized, crossover clinical trials, ASTX727-01-B, which included 80 adult patients with MDS (IPSS Intermediate-1, Intermediate-2, or high-risk groups) or CMML, and ASTX727-02, which included 133 adult patients with MDS or CMML, including all FAB subtypes and IPSS Intermediate-1, Intermediate-2, or high-risk groups. In these two trials, patients were randomized 1:1 to receive INQOVI® orally in cycle 1 and Decitabine 20 mg/m2 intravenously in cycle 2 or the reverse order. Both oral INQOVI® and intravenous Decitabine were administered once daily on days 1 through 5 of a 28-day cycle. Starting with cycle 3, all patients received INQOVI® orally once daily on days 1 through 5 of each 28-day cycle, until disease progression or unacceptable toxicity. Both trials provided comparison of exposure and safety in the first two cycles between oral INQOVI® and IV Decitabine and description of disease response with INQOVI®. Comparison of disease response between the INQOVI® and IV Decitabine was not possible because all patients received INQOVI® starting from Cycle 3. The Primary endpoint was total 5-day AUC exposures of Decitabine following INQOVI® therapy compared with IV Decitabine, as measured across the first 2 cycles. Secondary endpoints included safety assessments, pharmacodynamic measurements, clinical responses, RBC transfusion independence, Leukemia-free survival, and Overall Survival.
ASTX727-01-B trial which included 80 patients demonstrated a Complete Response (CR) rate of 18% and median duration of CR of 8.7 months. Among the 41 patients who were dependent on RBC and/or platelet transfusions at baseline, 49% became transfusion independent during any consecutive 56-day post-baseline period. Of the 39 patients who were independent of both RBC and platelet transfusions at baseline, 64% remained transfusion independent during any consecutive 56-day post-baseline period.
ASTX727-02 trial, which included 133 patients, demonstrated a 99% geometric mean ratio of the 5-day cumulative Decitabine AUC following 5 consecutive once daily doses of the oral combination therapy, versus that of IV Decitabine, with a 90% Confidence interval between 93% and 106%. This confirmed equivalence of oral INQOVI® and IV Decitabine. Efficacy results demonstrated that 21% of patients achieved CR, and median duration of CR was 7.5 months. Among the 57 patients who were dependent on RBC and/or platelet transfusions at baseline, 53% became transfusion independent during any 56-day post-baseline period. Of the 76 patients who were independent of both RBC and platelet transfusions at baseline, 63% remained transfusion independent during any 56-day post-baseline period. The most common Adverse Events related to INQOVI® included fatigue, rash, dizziness, headaches, anorexia, nausea, diarrhea, constipation, mucositis, hemorrhage, myalgia, arthralgia, febrile neutropenia, and transaminase elevation.
It was concluded that INQOVI® which is a fixed-dose combination of Cedazuridine and Decitabine is a new treatment option for patients with MDS and CMML, and is an oral hypomethylating agent alternative to IV Decitabine.
https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-oral-combination-decitabine-and-cedazuridine-myelodysplastic-syndromes
FDA Approves IO in Combination with Targeted Therapies for BRAF Positive Advanced Melanoma
SUMMARY: The FDA on July 30, 2020, approved TECENTRIQ® (Atezolizumab), in combination with COTELLIC® (Cobimetinib) and ZELBORAF® (Vemurafenib), for patients with BRAF V600 mutation-positive unresectable or metastatic melanoma. It is estimated that in the US, approximately 100,350 new cases of melanoma will be diagnosed in 2020 and approximately 6,850 patients are expected to die of the disease. The incidence of melanoma has been on the rise for the past three decades. Surgical resection with a curative intent is the standard of care for patients with early stage melanoma, with a 5-year survival rate of 98% for Stage I disease and 90% for Stage II disease. Patients with locally advanced or metastatic melanoma historically have had poor outcomes. With the development and availability of immune checkpoint inhibitors and BRAF and MEK inhibitors, this patient group now has significantly improved outcomes.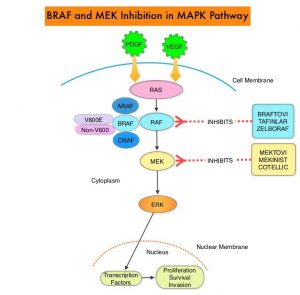
The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been detected in 6-8% of all malignancies. The most common BRAF mutation in melanoma is at the V600E/K site and is detected in approximately 50% of melanomas, and result in constitutive activation of the MAPK pathway.
ZELBORAF® (Vemurafenib), a selective oral inhibitor of mutated BRAF, demonstrated significant improvement in Progression Free Survival (PFS) and Overall Survival (OS), compared to Dacarbazine. Squamous cell carcinomas were seen in about 6% of the patients treated with BRAF inhibitors. Paradoxical activation of the MAPK pathway in cells without a BRAF mutation has been implicated in the emergence of drug resistance and increased incidence of BRAF-inhibitor induced skin tumors. MEK gene is downstream from RAF in the MAPK pathway. The addition of a selective inhibitor of MEK gene such as COTELLIC® (Cobimetinib) to a BRAF inhibitor such as ZELBORAF® has addressed some of these limitations, in previously published studies, with improvement in Objective Response Rates (ORR) and decrease in the incidence of cutaneous secondary cancers. coBRIM is a multicenter, randomized, Phase III study in which the efficacy and safety of COTELLIC® combined with ZELBORAF®, was evaluated in previously untreated patients, with advanced BRAF-mutated melanoma. The final analysis of this trial evaluated the 5-year survival data, and the OS was over 30% in patients who received the combination therapy, with a Complete Response (CR) rate was about 20%.
TECENTRIQ® (Atezolizumab) is an anti PD-L1 monoclonal antibody, designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors. PD-L1 inhibition may prevent T-cell deactivation and further enable the activation of T cells. The 5 year OS among patients receiving PD1 targeted immunotherapy is about 34%, with a median OS of 17-20 months. With the approval of multiple therapeutic options for the management of patients with BRAF-mutant melanoma, treatment decisions have become increasingly complex. In patients with limited disease burden, immunotherapy with checkpoint inhibitors is favored by most clinicians, based on the long term data supporting the durability of responses with immunotherapies, but response rates are lower. On the contrary, BRAF-targeted agents are utilized in patients with extensive, symptomatic disease and active brain metastases, as the response rates are higher but are short lived. The optimal sequence of these therapeutic strategies in order to improve long-term patient outcome, has remained unclear.
Preclinical studies suggested that combining these two targeted therapies with a checkpoint inhibitor might overcome the limitations of each class and potentially lead to more durable responses. The safety and efficacy of combining TECENTRIQ® with COTELLIC® (MEK inhibitor) and ZELBORAF® (BRAF inhibitor), in patients with BRAFV600-mutated metastatic melanoma, was evaluated in a Phase I study, with promising results, and a 28-day run-in period with COTELLIC® and ZELBORAF® was associated with an increase in proliferating CD4+ T-helper cells, without increasing the T-regulatory cells (Tregs). Tumor cells use Tregs as a shield to protect themselves against anti-tumor immune response and Tregs remain a hurdle in achieving the complete potential of anti-cancer therapies including immunotherapy. The aim of IMspire 150 trial was to determine if combining checkpoint inhibitor with two targeted therapies would improve efficacy.
IMspire150 is a pivotal, placebo-controlled, international, multicenter, double-blinded, Phase III trial, in which 514 treatment-naive patients with Stage IIIc and Stage IV, BRAF V600–mutant malignant melanoma were enrolled. Patients were randomly assigned 1:1 to treatment with the doublet combination or the triplet therapy. Doublet therapy given to the control group of patients consisted of ZELBORAF® 960 mg orally twice daily plus COTELLIC® at 60 mg orally, on days 1 to 21 of a 28 day cycle. In the experimental or triplet therapy group, there was a 28-day run-in with ZELBORAF® plus COTELLIC® alone, dosed similar to the control group (cycle 1), following which patients received TECENTRIQ® 840 mg IV on Days 1 and 15 of each 28 day cycle starting cycle 2, in combination with ZELBORAF® at a lower dose of 720 mg orally twice daily and COTELLIC® 60 mg orally once daily. Treatment was continued until disease progression, or unacceptable toxicity. Both treatment groups were well balanced, median patient age was 54 years, 58% were male and 94% of patients had Stage IV disease. The Primary endpoint was investigator-assessed Progression Free Survival (PFS). Secondary end points included Objective Response Rates (ORR), Duration of Response (DOR), and Overall Survival (OS).
The combination of immunotherapy with targeted therapies was significantly superior to targeted therapies alone. At a median follow up of 18.9 months, the median PFS with the triplet combination was 15.1 months versus 10.6 months with the doublet therapy (HR=0.78; P=0.025). This represented a 22% reduction in the risk of disease progression. This benefit was observed across all subgroups including age, disease burden, LDH level, and extent of tumor involvement by organ site. Although Objective Response Rates were similar in both treatment groups, the median Duration of Response was 21.0 months with triplet combination versus 12.6 months for the doublet therapy. The OS data were not mature at the time of this analysis, but interim analysis however showed a median OS of 28.8 months with the triplet combination versus 25.1 months with doublet therapy. Both treatment groups had comparable toxicities. Among those patients receiving triplet combination, the most common toxicities were rash, fever, fatigue, nausea, pruritus, stomatitis, musculoskeletal pain, hepatotoxicity, edema, hypothyroidism, and photosensitivity.
It was concluded that in treatment-naive patients with advanced BRAF V600-mutant malignant melanoma, TECENTRIQ® in combination with ZELBORAF® and COTELLIC® significantly and clinically improved Progression Free Survival, when compared to placebo in combination with ZELBORAF® and COTELLIC®.
Evaluation of atezolizumab (A), cobimetinib (C), and vemurafenib (V) in previously untreated patients with BRAFV600 mutation-positive advanced melanoma: Primary results from the phase 3 IMspire150 trial. McArthur GA, Stroyakovskiy D, Gogas H, et al. Presented at: the 2020 AACR Annual Virtual Meeting I; April 27-28, 2020. Abstract CT012.
TECENTRIQ®
The FDA on July 30, 2020 approved TECENTRIQ® (Atezolizumab) in combination with COTELLIC® (Cobimetinib) and ZELBORAF® (Vemurafenib) for patients with BRAF V600 mutation-positive unresectable or metastatic melanoma. TECENTRIQ® along with COTELLIC® and ZELBORAF® are products of Genentech, Inc.
TECARTUS®
The FDA on July 24, 2020 granted accelerated approval to TECARTUS® (brexucabtagene autoleucel), a CD19-directed genetically modified Autologous T cell immunotherapy, for the treatment of adult patients with relapsed or refractory Mantle Cell Lymphoma (MCL). TECARTUS® is a product of Kite Pharma, a subsidiary of Gilead Sciences.
INQOVI® (Decitabine and Cedazuridine)
The FDA on July 7, 2020 approved INQOVI® for adult patients with MyeloDysplastic Syndromes (MDS) including the following:
1) Previously treated and untreated, de novo and secondary MDS with the following French-American-British subtypes (Refractory Anemia, Refractory Anemia with Ringed Sideroblasts, Refractory Anemia with excess blasts, and Chronic MyeloMonocytic Leukemia [CMML])
2) Intermediate-1, Intermediate-2, and high-risk International Prognostic Scoring System groups.
INQOVI® is a product of Astex Pharmaceuticals, Inc.
The Transition to Biosimilars: Managing Payor Challenges
Written by Dr. Robert Rifkin | Sponsored by Mylan Pharmaceuticals
Biologic agents have long played a vital role in oncology. Not only does this class of agents represent the best of science, but it also accounts for a tremendous increase in spend of the healthcare dollar. As the field of biologic therapies advances, the biosimilarity exercise has become relevant. The premise of biosimilarity is to decrease healthcare costs and improve access to care.1
This premise was first established with the affordable care act in the Biologics Price Competition and Innovation Act (BCPIA).2 Since its inception, a new pathway for approval of biosimilars was tested and implemented. The 351 (K) regulatory pathway was first tested with biosimilar filgrastim (filgrastim–sndz), or Zarxio.3 Initially, upon product launch, a modest discount of 15% was employed. (15%) The uptake was slow; however, when the market adjusted and uptake accelerated, an approximate 30% discount was in play.
Several other biosimilars have now entered the supportive care space. Specifically, in the case of short acting filgrastim, there are now competitors Nivestym (Filgrastim-aafi) with several additional biosimilar filgrastims under development. Within the pegfilgrastim arena, the position of the originator, Neulasta, has now been challenged by 3 other long-acting filgrastims: Fulphila (pegfilgrastim-jmdb), Udenyca (pegfilgrastim-cbqv), and Ziextenzo (pegfilgrastim-bmez).4 These original, early supportive care biosimilars have helped to define the marketplace, test regulatory mechanisms, and dispel any myths regarding their adoption.4
Herceptin (trastuzumab) also faces competition as new biosimilars enter space. Multiple biosimilars have now launched in addition to the originator molecule, including: Herzuma (trastuzumab-pkrb), Kanjinti (trastuzumab-anns), Ogivri (trastuzumab-dkst), Ontruzant (trastuzumab-dttb), and Trazimera (trastuzumab-qyyp).5 For the trastuzumabs, the large number of biosimilar options provides both competition in the marketplace in addition to a potential new source of significant confusion with distribution, supply chain, and inventory management. It is relatively unlikely that any payor or formulary will carry all five biosimilar trastuzumabs currently available in addition to any other biosimilar options slated for release over the next year.
Additionally, the rituximab space has become increasingly complex. Beyond the originator molecule, we can now choice to use Truxima (ritiximab abbs), or Ruxience (rituximab-pvvr), and several more launches are anticipated. The space is further complicated by the availability of a subcutaneous form of Rituxan Hycela (rituximab/hyaluronidase human), the only biosimilar available in subcutaneous injection. Unsurprisingly, this has created some from payors as all labeled indications are not initially the same for each product. Moving forward, rituximab biosimilar labels will soon be equivalent, and competition will then drive the marketplace.
In the real world, there still exist very real barriers to adoption including a clinical, ease of use, and economic barriers. It is likely payors will interpose themselves into each one of these. Multiple biosimilars are now being approved for each originator molecule. This will ultimately result in a decline in cost. Payors and other stakeholders will then be faced with complicated decisions of maintaining the originator on the formulary, deleting it and placing a biosimilar in its place, or perhaps carrying two versions of the same molecule, with preference being given to one. Most likely, most formularies will carry originator in addition to one or more biosimilars concurrently depending on the provider and payer landscape. the originator and a preferred biosimilar concurrently.
Several articles have reviewed the concept of switching between the biosimilar and the originator, and to date no significant safety signals have arisen.6 The payor landscape is impacted by product availability and opportunities to switch. This demonstrated safety of switching has incrementally impacted the payor landscape. Pharmacy Benefit Managers (PBM) have also been interwoven into the payor conundrum, with discounts and rebates providing an additional layer of complexity.
Not only is the transition to biosimilars complicated for payors, but the transition must also include all stakeholders involved in the ultimate selection of a therapeutic biologic. Providers and patients need to be very well educated regarding the concept of biosimilarity. Other stakeholders, including pharmacists, advanced practice providers, nurses, and admixture technologists, must be thoroughly educated. The electronic health record also needs to be updated to reflect the increasing numbers of biosimilars now available – including the preferred therapeutic agents in each circumstance.
Payors and clinicians alike will need to develop biosimilar teams, drug contracting strategies with and without GPO’s, and a thorough evaluation of clinical economics for each biosimilar. Biosimilars will assume an increasingly important role in the delivery of cancer care and it is important to approach this from a patient journey point of view (Fig. 1).
Figure 1: Patient Journey7
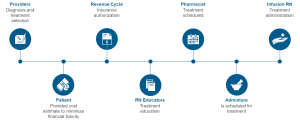
By tracing this from beginning to end, a formula for success may be developed.
The combination of clinical confidence, patient confidence, and operational excellence will be required to be sure that we are prepared for biosimilars and ensure patient access. The patient’s journey is complicated and increasingly influenced by the payor and other stakeholders . Providers, consideration of the revenue cycle, RN educators, pharmacists, admixture technologists, and infusion RNs all must play together to ensure the success of biosimilars. In alignment with the patient journey, diagnosis and treatment selection will be accomplished by the provider. This will be followed by insurance authorization, treatment education, treatment scheduling, treatment admixture, and finally the delivery of the drug to the well-educated patient. There are many potentials for success as well as failure (Fig. 2).
Figure 2: Drug Preparedness – Success vs. Failure7
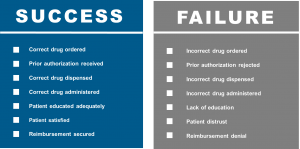
The cornerstone for all to succeed, as well as all of the affected stakeholders managing paired challenges, remains education. This cannot be overstated. Numerous websites have now appeared, both branded and unbranded, to help deliver biosimilar education. Such websites may be found at the FDA8 and the Center for Biosimilars.9
In conclusion, as all the stakeholders become thoroughly educated, the many challenges outlined above will continue to present themselves in real-time. The success and adoption of current and future biosimilars will continue to depend on the sound education of all stakeholders, including payors, in addition to improved cost savings and access. Biosimilar usage will be important to ensure long-term sustainability on the market, and as biosimilar uptake increases, healthcare cost reduction and improvements to care access may be achieved.
Sources
1. Pittman WL, Wern C, Glode AE. Review of Biosimilars and Their Potential Use in Oncology Treatment and Supportive Care in the United States.
2. https://www.fda.gov/media/78946/download
3. U.S. Food & Drug Administration. Zarxio (filgrastim-sndz) Approval Letter. Published online March 6, 2015. Accessed June 19, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/125553Orig1s000Approv.pdf
4. Rugo H, Rifkin RM, Deckerc P, Bair AH, Morgan G. Demystifying Biosimilars: Development, Regulation and Clinical Use. Future Oncology. 15(7):777-790, 2019
5. AmerisourceBergen. Approval and launch dates for US biosimilars. Published June 19, 2020. Accessed June 25, 2020. http://gabionline.net/Reports/Approval-and-launch-dates-for-US-biosimilars?ct=t%28GONL+V20F19-6%29&mc_cid=2821e641cc&mc_eid=%5BUNIQID%5D
6. Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching Reference Medications to Biosimilars: A Systematic Literature Review of Clinical Outcomes. Drugs 78(4): 463-78,2018.
7. Rifkin R, Busby L. Bringing Biosimilars to Community. Presented at McKesson Oncology University; 2019.
8. www.fda.gov
9. www.centerforbiosimilars.com/
ENHERTU® Improves Overall Survival in Previously Treated HER2-Positive Gastric Cancer
SUMMARY: The American Cancer Society estimates that in the US, about 27,600 new cases of Gastric cancer will be diagnosed in 2020 and about 11,010 people will die of the disease. It is one of the leading causes of cancer-related deaths in the world. Several hereditary syndromes such as Hereditary Diffuse Gastric Cancer (HDGC), Lynch syndrome (Hereditary Nonpolyposis Colorectal Cancer) and Familial Adenomatous Polyposis (FAP) have been associated with a predisposition for stomach cancer. Additionally, one of the strongest risk factor for Gastric adenocarcinoma is infection with Helicobacter pylori (H.pylori), which is a gram-negative, spiral-shaped microaerophilic bacterium.
The Human Epidermal growth factor Receptor (HER) or erbB family of receptors, consist of HER1, HER2, HER3 and HER4. Approximately 15-20% of advanced Gastric and GastroEsophageal (GE) junction cancers, overexpress or have amplification of the HER2 oncogene. These patients often receive first line treatment with a combination of chemotherapy plus anti-HER2 antibody, Trastuzumab, as there is Overall Survival (OS) benefit with this combination regimen. Upon progression, Paclitaxel plus CYRAMZA® (Ramucirumab), an anti-VEGFR-2 antibody is recommended as second-line therapy, regardless of HER2 expression, based on OS and Progression Free Survival (PFS) data for this combination regimen. Trifluridine-tipiracil (LONSURF®) and Immune Checkpoint Inhibitors are treatment options for later lines of therapy and are associated with minimal prolongation in OS. Unlike treatment in metastatic breast cancer, re-treatment with Trastuzumab in combination with various different chemotherapy agents has not shown survival benefit in Gastric cancer. Further, Antibody-Drug Conjugate such as KADCYLA® (ado-trastuzumab emtansine), did not prolong median OS or improve Response Rates compared to chemotherapy, in patients with Gastric cancer who had progressed during or after treatment with Trastuzumab.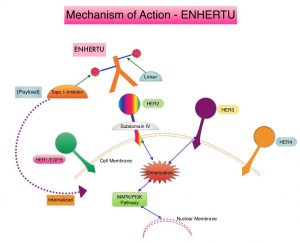
ENHERTU® (Trastuzumab Deruxtecan) is an Antibody-Drug Conjugate (ADC) composed of a humanized monoclonal antibody specifically targeting HER2, with the amino acid sequence similar to Trastuzumab, a cleavable tetrapeptide-based linker, and a potent cytotoxic Topoisomerase I inhibitor as the cytotoxic drug (payload). ENHERTU® has a favorable pharmacokinetic profile and the tetrapeptide-based linker is stable in the plasma and is selectively cleaved by cathepsins that are up-regulated in tumor cells. Unlike KADCYLA®, ENHERTU® has a higher drug-to-antibody ratio (8 versus 4), released payload easily crosses the cell membrane with resulting potent cytotoxic effect on neighboring tumor cells regardless of target expression, and the released cytotoxic agent (payload) has a short half-life, minimizing systemic exposure.
DESTINY-Gastric01 is an open-label, randomized, multicenter, Phase II trial in which ENHERTU® was compared with chemotherapy in patients with HER2-positive advanced Gastric cancer. In this study 187 patients were randomly assigned in a 2:1 ratio to receive either ENHERTU® (N=125) or the treating physician’s choice of Irinotecan or Paclitaxel (N=62). Eligible patients had HER2-expressing advanced Gastric cancer or GastroEsophageal junction adenocarcinoma that had progressed after the receipt of at least two previous systemic therapies, which included a Fluoropyrimidine, a Platinum agent, and Trastuzumab (or approved biosimilar agent). Patients in the ENHERTU® group received the drug at a dose of 6.4 mg/kg as IV infusion every 3 weeks, whereas the physician’s choice group received either Irinotecan monotherapy 150 mg/m2 IV every 2 weeks, or Paclitaxel monotherapy 80 mg/m2 IV on days 1, 8, and 15 every 4 weeks. HER2 levels were documented as high if the score was 3+ on IHC, or 2+ on IHC with positive results on FISH, and documented as low if the score was 2+ on IHC with negative results on FISH, or a score of 1+ on IHC (negative). Treatment was continued until disease progression or unacceptable toxicities. Both treatment groups were well balanced. Approximately 72% of the patients had previously received CYRAMZA® (Ramucirumab), and 86% had received Taxanes. The median time since the last administration of Trastuzumab was 6.2 months. The Primary end point was the Objective Response Rate (ORR), according to Independent Central Review. Secondary end points included Overall Survival (OS), response duration, Progression Free Survival, and safety. The primary cohort consisted of patients with high-level HER2-positive disease, and was the focus of this analysis.
Treatment with ENHERTU® resulted in an ORR of 51%, compared to 14% in the physician’s choice group (P<0.001), according to Independent Central Review. An ORR lasting 4 weeks or more occurred in 43% of patients in the ENHERTU® group, as compared with 12% in the physician’s choice group. More than 80% of patients receiving ENHERTU® had a reduction in tumor size, compared with approximately half the patients receiving physician’s choice of chemotherapy. The median duration of confirmed objective response was 11.3 months in the ENHERTU® group, compared with 3.9 months in the physician’s choice group. Treatment with ENHERTU® resulted in a higher percentage of patients with confirmed disease control (86%), than physician’s choice of chemotherapy (62%). The ORR was higher among those with a HER2 score of 3+ on IHC, than among those with an IHC score of 2+ with positive results on FISH (58% versus 29%).
The Overall Survival was significantly longer in the ENHERTU® group compared to the physician’s choice group (median 12.5 months versus 8.4 months; HR=0.59; P=0.01). The estimated OS at 6 months was 80% in the ENHERTU® group and 66% in the physician’s choice group and at 12 months was 52% and 29%, respectively. In a prespecified subgroup analysis, OS benefit was greater with ENHERTU® compared to physician’s choice of chemotherapy, across most subgroups. The median PFS was 5.6 months in the ENHERTU® group and 3.5 months in the physician’s choice group (HR=0.47). The most common adverse events of Grade 3 or higher were cytopenias. ENHERTU® related Interstitial Lung Disease or pneumonitis was noted in 10% of patients and most events were Grade 1 or 2. Decrease in left ventricular ejection fraction or heart failure was not observed in either treatment groups.
It was concluded that treatment with ENHERTU® resulted in significant improvements in Objective Response Rates and Overall Survival, as compared with standard therapies, among patients with HER2-positive advanced Gastric or GastroEsophageal junction cancer. This benefit was seen even in patients who had disease progression while on Trastuzumab containing regimens.
Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. Shitara K, Bang Y-J, Iwasa S, et al. for the DESTINY-Gastric01 Investigators. N Engl J Med 2020; 382:2419-2430