
The FDA on March 5, 2021 granted accelerated approval to YESCARTA® for adult patients with Relapsed or Refractory Follicular Lymphoma (FL) after two or more lines of systemic therapy. YESCARTA® is a product of Kite Pharma, Inc.
Author: RR
PEPAXTO® (Melphalan flufenamide)
The FDA on February 26, 2021, granted accelerated approval to PEPAXTO® in combination with Dexamethasone for adult patients with relapsed or refractory multiple myeloma who have received at least four prior lines of therapy and whose disease is refractory to at least one Proteasome Inhibitor, one immunomodulatory agent, and one CD-38 directed monoclonal antibody. PEPAXTO® is a product of Oncopeptides, Inc.
Worse Outcomes from COVID-19 Infection in Lymphoma Patients Treated with Anti-CD20 Monoclonal Antibodies
SUMMARY: The SARS-CoV-2 Coronavirus (COVID-19) induced pandemic first identified in December 2019 in Wuhan, China, has contributed to significant mortality and morbidity in the US, and the number of infections, continue to exponentially increase worldwide. Majority of the patients present with treatment-resistant pyrexia and respiratory insufficiency, with some of these patients progressing to a more severe systemic disease and multiple organ dysfunction.
Patients with lymphoproliferative disorders may be immune deficient due to their underlying disease or due to the therapies they receive, which in turn can increase the incidence and severity of infections. Patients with Non Hodgkin Lymphoma are often treated with CD20 targeted, B-cell depleting monoclonal antibodies such as RITUXAN® (Rituximab) or GAZYVA® (Obinutuzumab), as they were shown to improve survival among patients with B-cell Non-Hodgkin Lymphoma. Depleting B cells dampens the body’s ability to generate antibody responses to new pathogens, which may impact the clinical course of COVID-19. The authors in this study analyzed the clinical course of COVID-19 infection in lymphoma patients, and characterized the determinants of worse outcomes.
It has been shown in several studies and registries that patients with hematologic malignancies including lymphomas have a higher incidence of death from COVID-19 compared with other types of cancer. Additional risk factors for COVID-19-related mortality include older age and relapsed or refractory disease. To better understand the risk factors associated with worse outcomes from COVID-19 in this patient population, the authors conducted a retrospective study of 111 patients with lymphoma hospitalized for COVID-19 at any of the 16 French hospitals during March and April 2020. The researchers specifically focused on identifying factors associated with prolonged hospital stay (longer than 30 days), or hospitalization for recurrent symptoms for more than 30 days and death, and used length of hospital stay as a proxy for persistent COVID-19 infection. Study patients included those formerly treated for lymphoma, those currently undergoing treatment, or had no treatment.
Of the 111 patients included in this study, 57% (N=63) had previously received B-cell-depleting therapy. The most common type of lymphoma was Diffuse Large B-Cell Lymphoma. Twenty nine percent (29%) of all patients required a prolonged hospital stay (longer than 30 days) due to severe COVID-19 symptoms and persistent disease. The median age of patients with persistent COVID-19 was 64 years and 63% were male. More than two-thirds (69%) had at least one significant comorbidity. None of the patients with T-cell lymphoma included in the study (N=8) experienced persistent COVID-19 infection.
At a median follow-up of 191 days, the 6-month Overall Survival for the entire cohort was 69%. Older age (70 years and over) as well as relapsed/refractory disease were both associated with worse survival and prolonged hospital stays. After adjusting for age, comorbidities, and the presence of relapsed/refractory disease, the researchers noted that receipt of B-cell-depleting treatment within the previous 12 months nearly doubled the likelihood of a prolonged hospital stay and more than doubled the risk of death. After 1 month, 41% of patients who received anti-CD20 monoclonal antibodies were still hospitalized for COVID-19 versus 13% not treated with those antibodies.
The authors concluded that standardized guidelines on the use of anti-CD20 therapies are needed to help us make decisions during the COVID-19 pandemic, and convalescent plasma may be a treatment consideration for B-cell-depleted patients with persistent COVID-19. Patients who recently received B-cell depleting therapies and have COVID-19 should be closely monitored. Additionally, the efficacy and timing of vaccination in this particular population needs further study.
High incidence of persistent COVID-19 among patients with lymphoma treated with B-cell depleting immunotherapy. Lamure S, Dulery R, Delord M, et al. AACR Virtual Meeting: COVID-19 and Cancer. Abstract S09-02. Presented on February 5, 2021.
FDA Approves LORBRENA® for Advanced ALK-Positive Lung Cancer
SUMMARY: The FDA on March 3, 2021, granted regular approval to LORBRENA® (Lorlatinib) for patients with metastatic Non Small Cell Lung Cancer (NSCLC) whose tumors are Anaplastic Lymphoma Kinase (ALK)-positive, as detected by an FDA-approved test. The FDA also approved the Ventana ALK (D5F3) CDx Assay (Ventana Medical Systems, Inc.) as a companion diagnostic for LORBRENA®. Lung cancer is the leading cause of cancer death in both men and women, and accounts for about 14% of all new cancers and 25% of all cancer deaths. The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The discovery of chromosomal rearrangements of the Anaplastic Lymphoma Kinase (ALK) gene in some patients with advanced NSCLC and adenocarcinoma histology, and their sensitivity to ALK inhibitors, paved the way to the development of small-molecule ALK Tyrosine Kinase Inhibitors. It has become clear that appropriate, molecularly targeted therapy for tumors with a molecular abnormality, results in the best outcomes. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8%, as well as other mutations in BRAF, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive, and the presence of two simultaneous mutations, are rare.
ALK inhibitors include first-generation XALKORI® (Crizotinib) and second-generation ALK inhibitors such as ZYKADIA® (Ceritinib), ALECENSA® (Alectinib) and ALUNBRIG® (Brigatinib). Despite the improved efficacy of second-generation ALK inhibitors, recurrent disease due to drug resistance including CNS disease progression, can still develop.
LORBRENA® is a novel third-generation ALK inhibitor that is more potent than second-generation inhibitors, and has the broadest coverage of ALK resistance mutations that have been identified. LORBRENA® crosses the blood-brain barrier and has marked intracranial activity in previously treated patients with baseline CNS disease, including leptomeningeal disease. LORBRENA® received accelerated approval by the FDA in November 2018 for the second or third-line treatment of ALK-positive metastatic NSCLC. However, the efficacy of LORBRENA®, as compared with that of XALKORI®, as first line treatment for advanced ALK-positive NSCLC, has been unclear.
The CROWN trial is a global, open label, randomized, Phase 3 study, in which LORBRENA® was compared with XALKORI®, in patients with previously untreated ALK-positive advanced NSCLC. In this study, 296 treatment naïve advanced NSCLC patients were randomly assigned 1:1 to receive LORBRENA® 100 mg orally once daily (N=149) or XALKORI® 250 mg orally twice daily (N=147) in cycles of 28 days. Treatment was continued until disease progression or unacceptable toxic effects. Eligible patients were required to have ALK-positive tumors detected by the Ventana ALK (D5F3) CDx assay. Patients with asymptomatic treated or untreated CNS metastases were eligible and had to have at least one extracranial measurable target lesion that had not been previously irradiated. Patients were stratified according to the presence of brain metastases and ethnic group (Asian or non-Asian) and crossover between the treatment groups was not permitted. The Primary end point was Progression Free Survival (PFS) as assessed by Blinded Independent Central Review (BICR). Secondary end points included independently assessed Objective Response Rate (ORR) and intracranial response.
At a planned interim analysis, treatment with LORBRENA® resulted in statistically significant and clinically meaningful improvement in PFS as assessed by BICR, with a Hazard Ratio of 0.28 (P<0.001), corresponding to a 72% reduction in the risk of disease progression or death. The median PFS was not estimable in the LORBRENA® arm and was 9.3 months for those treated with XALKORI®. The percentage of patients who were alive without disease progression at 12 months was 78% in the LORBRENA® group and 39% in the XALKORI® group, and the Hazard Ratio favored LORBRENA® over XALKORI® across all prespecified patient subgroups. The Overall Survival data were immature at the PFS analysis.
The confirmed ORR was 76% with LORBRENA® and 58% with XALKORI®. About 70% of the patients who received LORBRENA® and 27% of those who received XALKORI® had a response that lasted at least 12 months. Additionally, treatment with LORBRENA® was associated with increased intracranial activity compared with XALKORI®. Among patients presenting with measurable brain metastases, the intracranial ORR was 82% with LORBRENA® and 23% with XALKORI®, with a intracranial Complete Response rate of 71% and 8%, respectively. The duration of intracranial response was 12 months or more in 79% and 0% of patients in the LORBRENA® and XALKORI® groups, respectively. The most common adverse events with LORBRENA® were hyperlipidemia, edema, weight gain, peripheral neuropathy, and cognitive effects.
It was concluded that treatment LORBRENA® resulted in a significantly longer Progression Free Survival and a higher frequency of intracranial response, compared to XALKORI®, among patients with previously untreated advanced ALK-positive NSCLC.
First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. Shaw AT, Bauer TM, de Marinis F, et al. N Engl J Med 2020; 383:2018-2029.
Role of Aspirin in Colorectal Cancer Prevention and Mortality
SUMMARY: Colorectal Cancer (CRC) is the third leading cause of cancer-related deaths in men and women in the United States. The American Cancer Society estimates that approximately 149,500 new cases of CRC will be diagnosed in the United States in 2021 and about 52,980 patients will die of the disease. The lifetime risk of developing CRC is about 1 in 23. The majority of CRC cases (about 75 %) are sporadic whereas the remaining 25 % of the patients have family histories of the disease. Only 5-6 % of patients with CRC with a family history background are due to inherited mutations in major CRC genes, while the rest are the result of accumulation of both genetic mutations and epigenetic modifications of several genes. Colorectal Cancer is a heterogeneous disease classified by its genetics, and even though the overall death rate has continued to drop, deaths from CRC among people younger than 55 years have increased 1% per year from 2008 to 2017, with 12% of CRC cases diagnosed in people under age 50.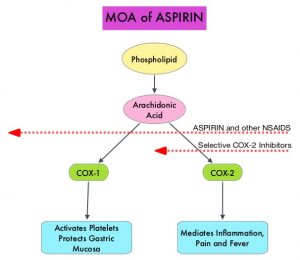
Aspirin (AcetylSalicylic Acid) has been studied as a chemopreventive agent for several decades and the temporal relationship between systemic inflammation and cancer has been a topic of ongoing investigation. The US Preventive Services Task Force (USPSTF) found adequate evidence that Aspirin use reduces the incidence of CRC in adults after 5-10 years of use, and recommends initiating low-dose Aspirin use for the primary prevention of CardioVascular Disease (CVD) and CRC, in adults aged 50-69 years, who have a 10% or greater 10-year CVD risk, are not at increased risk for bleeding, have a life expectancy of at least 10 years, and are willing to take low-dose Aspirin daily for at least 10 years.
The molecular mechanisms underlying Aspirin’s chemoprevention effects as well as the dose, duration, and timing of Aspirin chemoprevention have remained unclear. More recent data suggests that platelets may play a role in tumorigenesis as well, through the release of angiogenic and growth factors due to overexpression of COX-2. Daily low dose Aspirin inhibits COX-1 and COX-2. It is postulated that Aspirin also works by COX-independent mechanisms such as, the inhibition of NF-kB and Wnt/ β-catenin signaling, which may play a role in its chemopreventive properties.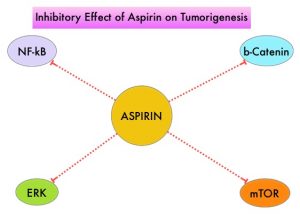
Two recently published studies have provided new information on the association between regular aspirin use, and CRC incidence.
In the first study by Zhang and colleagues, the authors addressed the use of aspirin for the primary prevention of CRC and explored the dosing, timing and duration of Aspirin intake, to reduce the incidence of CRC. The authors derived data from two large US cohort studies, The Nurses’ Health Study (January 1980-June 2014) and the Health Professionals Follow-up Study (January 1986-January 2014). These two studies provided data on the use of Aspirin by more than 94,500 participants over a period of 35 years, providing a unique opportunity to evaluate the chemopreventive benefit of Aspirin on CRC. Colorectal cancer incidence was approximately 10% lower amongst individuals reporting regular Aspirin use immediate 10 years or more earlier before follow up started, with the lowest average dose studied (23-70 mg/day) appearing as effective as higher doses. However, when the period of Aspirin use was between 5 and 10 years earlier, a smaller reduction in CRC incidence was noted amongst those taking aspirin, but the greatest reduction was noted among those taking the highest dose. This study suggested that the benefit necessitates at least 6-10 years and most clearly after approximately 10 years since initiation of Aspirin. Both remote use and use within the previous 10 years contributed independently to decreased risk, though a lower dose may be required for a benefit with longer term use.
Figueiredo and colleagues studied whether the use of Aspirin prior to or after a diagnosis of CRC, affects subsequent cancer-related mortality. They utilized data from men and women enrolled in the American Cancer Society’s Cancer Prevention Study-II (CPS-II) Nutrition Cohort who were cancer-free at the baseline (year 1992/1993), and diagnosed with CRC during follow up through 2015. They compared CRC-specific mortality amongst long-term regular Aspirin users (defined as 15 or more times per month) and non-users prior to and after diagnosis. Long-term regular use of Aspirin before diagnosis was associated with lower CRC mortality. Regular use of Aspirin following diagnosis was not statistically significantly associated with risk of CRC-specific mortality overall, although participants who began regular Aspirin use, only after their diagnosis, were at lower risk, than participants who did not use Aspirin at both the pre-and post-diagnosis periods. Further, long-term Aspirin use before diagnosis was also associated with lower risk of distant metastases. This study suggested that long-term Aspirin use before a diagnosis of non-metastatic CRC may be associated with lower CRC-specific mortality after diagnosis, implying possible inhibition of micro-metastases before diagnosis.
Taken together, the study by Zhang et al. suggested that even a relatively small dose of Aspirin taken regularly for 2-5 years during the middle years of life might reduce the risk of colorectal cancer 10 years or so later, whereas the study by Figueiredo and colleagues suggested that Aspirin, taken prior to (or started after) a diagnosis of colorectal cancer, reduced subsequent metastatic spread and cancer related mortality. Even though these two new studies support the anti-cancer effect of Aspirin, additional consistent information may be required before widespread role of Aspirin for primary prevention of cancer is embraced.
Timing of aspirin use in colorectal cancer chemoprevention: a prospective cohort study. Zhang Y, Chan AT, Meyerhardt JA, et al. J Natl Cancer Inst 2021; https://doi.org/10.1093/jnci/djab009
Associations of aspirin and non-aspirin non-steroidal anti-inflammatory drugs with colorectal cancer mortality after diagnosis. Figueiredo JC, Jacobs EJ, Newton CC, et al. J Natl Cancer Inst 2021; https://doi.org/10.1093/jnci/djab008
PADCEV® Prolongs Overall Survival in Metastatic Urothelial Carcinoma
SUMMARY: The American Cancer Society estimates that in 2021, approximately 83,730 new cases of Bladder Cancer will be diagnosed and 17,200 patients will die of the disease. Patients with urothelial carcinoma are currently treated in the first line setting with a Platinum based chemotherapy regimen and a checkpoint Inhibitor (PD-1 or PD-L1 inhibitor) in the second line setting. Treatment options for patients who progress after first and second line therapies are limited, with poor outcomes. The response rates with standard chemotherapy in this patient population, is about 10%.
PADCEV® (Enfortumab vedotin-ejfv) is an Antibody-Drug Conjugate (ADC) that targets Nectin-4, a cell adhesion molecule highly expressed in urothelial cancers and other solid tumors. Nectin-4 has been implicated in tumor cell growth and proliferation. Following binding to Nectin-4 on the cell surface, PADCEV® becomes internalized and is processed by lysosomes, with the liberation of its cytotoxic payload, Monomethyl auristatin E, which in turn disrupts microtubule assembly, leading to cell cycle arrest and apoptosis. The FDA in 2019 granted accelerated approval to PADCEV® for adult patients with locally advanced or metastatic urothelial cancer, who have previously received a Programmed Death receptor-1 (PD-1) or Programmed Death-Ligand1 (PD-L1) inhibitor, and a Platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced or metastatic setting. This approval was based on the results from the pivotal Phase II EV-201 study which concluded that treatment with PADCEV® demonstrated a clinically meaningful 44% Objective Response Rate (ORR) in this patient group. EV-301 study was designed to confirm the clinical benefit of PADCEV® as compared with standard chemotherapy, by assessing Overall Survival in patients with advanced urothelial carcinoma, who had previously received treatment.
EV-301 is a global, open-label, randomized, Phase III trial, that evaluated the efficacy of PADCEV®, as compared with chemotherapy, in patients with locally advanced or metastatic urothelial carcinoma, who had previously received treatment with a platinum-based chemotherapy and a PD-1 or PD-L1 inhibitor. A total of 608 patients were randomly assigned 1:1 to receive either PADCEV® 1.25 mg/kg IV on days 1, 8, and 15 of a 28-day cycle (N=301), or investigator choice of chemotherapy (Docetaxel 75 mg/m2 IV, Paclitaxel 175 mg/m2 IV, or Vinflunine 320 mg/m2 IV, administered on day 1 of a 21-day cycle (N=307). Both treatment groups were well balanced. The median patient age was 68 years, about 79% of patients had visceral metastases, and 69% of patients had no response to previous treatment with checkpoint inhibitor. The Primary end point was Overall Survival (OS). Secondary end points included Progression Free Survival (PFS) and Overall Response Rate (ORR) and Safety. At the prespecified interim analysis, the median follow up was 11.1 months.
The median OS was 12.88 months in the PADCEV® group versus 8.97 months in patients treated with chemotherapy (H= 0.70; P=0.001), suggesting a 30% reduction in the risk of death with PADCEV®. The PFS was also longer in the PADCEV® group, and was 5.6 months versus 3.7 months with chemotherapy (HR=0.62; P<0.001). The confirmed ORR was higher in the PADCEV® group than in the chemotherapy group (40.6% versus 17.9%; P<0.001), and a Complete Response was observed in 4.9% of the patients in the PADCEV® group and in 2.7% of the patients in the chemotherapy group. Disease Control Rate was 71.9% and 53.4%, respectively (P<0.001). Treatment-related Adverse Events were similar in the two treatment groups.
The authors concluded that treatment with PADCEV® resulted in significantly longer Overall Survival, Progression Free Survival and a higher Overall Response Rate, than standard chemotherapy, in patients with locally advanced or metastatic urothelial carcinoma, who had previously received Platinum-based treatment and a PD-1 or PD-L1 inhibitor.
Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. Powles T, Rosenberg JE, Sonpavde GP, et al. N Engl J Med 2021; DOI: 10.1056/NEJMoa2035807
MARGENZA® Superior to Trastuzumab in Heavily Pretreated HER2-Positive Breast cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 284,200 new cases of breast cancer will be diagnosed in 2021 and about 44,130 individuals will die of the disease, largely due to metastatic recurrence. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. Patients with HER2-positive metastatic breast cancer are often treated with anti-HER2 targeted therapy along with chemotherapy, irrespective of hormone receptor status, and this has resulted in significantly improved treatment outcomes. HER2 oncoprotein is also expressed by tumor cells in GastroEsophageal and other solid tumors.
HER2-targeted therapies include HERCEPTIN® (Trastuzumab), TYKERB® (Lapatinib), PERJETA® (Pertuzumab) and KADCYLA® (Ado-Trastuzumab Emtansine). Dual HER2 blockade with HERCEPTIN® and PERJETA®, given along with chemotherapy (with or without endocrine therapy), as first line treatment, in HER2 positive metastatic breast cancer patients, was shown to significantly improve Progression Free Survival (PFS) as well as Overall Survival (OS). The superior benefit with dual HER2 blockade has been attributed to differing mechanisms of action and synergistic interaction between HER2 targeted therapies. Patients progressing on Dual HER2 blockade often receive KADCYLA® which results in an Objective Response Rate (ORR) of 44% and a median PFS of 9.6 months, when administered after HERCEPTIN® and a taxane. There is however no standard treatment option for this patient population following progression on KADCYLA®.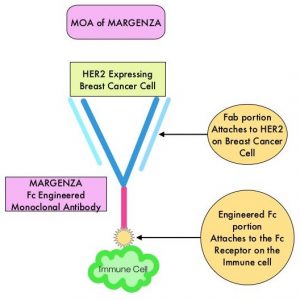
MARGENZA® (Margetuximab-cmkb) is an Fc-engineered, monoclonal antibody that binds to the HER2 oncoprotein with high specificity and affinity and inhibits tumor cell proliferation and survival, by mediating Antibody-Dependent Cellular Cytotoxicity (ADCC). It is postulated that the Fab portion of MARGENZA® has the same specificity and affinity to HER2 oncoprotein as Trastuzumab, with similar ability to disrupt signaling. However, the modified Fc region of MARGENZA®, which binds to Fc receptor expressing cells such as immune cells, has increased affinity for activating Fc receptor FCGR3A (CD16A) and decreased affinity for inhibitory Fc receptor FCGR2B (CD32B). These changes lead to greater ADCC and Natural Killer cell activation. Approximately 85% of individuals are CD16A-158F allele carriers.
The SOPHIA study is a randomized, multicenter, open-label, Phase III clinical trial, in which MARGENZA® plus chemotherapy was compared to Trastuzumab plus chemotherapy in patients with HER2-positive metastatic breast cancer, who have previously been treated with anti-HER2-targeted therapies. This study enrolled 536 patients who were randomized 1:1 to receive either MARGENZA® 15 mg/kg IV every three weeks (N=266) or Trastuzumab 6 mg/kg (8 mg/kg loading dose) IV every three weeks (N=270), in combination with Capecitabine, Eribulin, Gemcitabine or Vinorelbine, given at the standard doses. The median age was 56 years, all study patients had previously received Trastuzumab, all but one patient had previously received PERJETA® (Pertuzumab), and 91% of patients had previously received KADCYLA®. Patients were stratified by choice of chemotherapy, number of lines of therapy in the metastatic setting and number of metastatic sites. The dual Primary endpoints of the study were Progression Free Survival (PFS) by Blinded Independent Central Review (BICR) and Overall Survival (OS). Additional efficacy outcome measures included Objective Response Rate (ORR) and Duration of Response (DOR) assessed by BICR.
This study demonstrated a statistically significant 24% reduction in the risk of disease progression or death with MARGENZA® plus chemotherapy compared with Trastuzumab plus chemotherapy (HR= 0.76; P=0.03), with a median PFS of 5.8 months versus 4.9 months respectively. Treatment benefit was more pronounced in patients with CD16A genotypes containing a 158F allele (median PFS 6.9 versus 5.1 months, HR=0.68; P=0.005). The ORR for MARGENZA® plus chemotherapy was 22%, with a median Duration of Response of 6.1 months, compared to an ORR of 16% and median Duration of Response of 6.0 months for Trastuzumab plus chemotherapy. After the second planned interim analysis, the median OS was 21.6 months with MARGENZA® versus 19.8 months with Trastuzumab (HR= 0.89; P=0.33) and the ORR was 25% versus 14% respectively (P<0.001). The final Overall Survival (OS) analysis is expected in the second half of 2021. Safety was comparable in treatment groups, although the incidence of infusion-related reactions, mostly in cycle 1, was higher with MARGENZA® (13.3% versus 3.4%).
It was concluded that MARGENZA® in combination with chemotherapy significantly improved PFS, compared to Trastuzumab plus chemotherapy, in pretreated patients with HER2 positive metastatic breast cancer. MARGENZA® along with chemotherapy represents the newest treatment option for patients who have progressed on available HER2-directed therapies.
Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. Rugo HS, Im SA, Cardoso F, et al. for the SOPHIA Study Group. JAMA Oncol. Published online January 22, 2021. doi:10.1001/jamaoncol.2020.7932
2021 ASCO GU Cancers Symposium: Apalutamide and Abiraterone plus Prednisone Improves PFS in Chemo-Naive mCRPC Patients
SUMMARY: Prostate cancer is the most common cancer in American men with the exclusion of skin cancer, and 1 in 8 men will be diagnosed with prostate cancer during their lifetime. It is estimated that in the United States, about 248,530 new cases of prostate cancer will be diagnosed in 2021 and 34,130 men will die of the disease. The development and progression of prostate cancer is driven by androgens. Androgen Deprivation Therapy (ADT) or testosterone suppression has therefore been the cornerstone of treatment of advanced prostate cancer, and is the first treatment intervention. Androgen Deprivation Therapies have included bilateral orchiectomy or Gonadotropin Releasing Hormone (GnRH) analogues, with or without first generation Androgen Receptor (AR) inhibitors such as CASODEX® (Bicalutamide), NILANDRON® (Nilutamide) and EULEXIN® (Flutamide) or with second generation anti-androgen agents, which include ZYTIGA® (Abiraterone), XTANDI® (Enzalutamide), ERLEADA® (Apalutamide) and NUBEQA® (Darolutamide). Approximately 10-20% of patients with advanced Prostate cancer will progress to Castration Resistant Prostate Cancer (CRPC) within five years during ADT, and over 80% of these patients will have metastatic disease at the time of CRPC diagnosis. Among those patients without metastases at CRPC diagnosis, 33% are likely to develop metastases within two years. Progression to Castration Resistant Prostate Cancer (CRPC) often manifests itself with a rising PSA (Prostate Specific Antigen), and watchful waiting is often recommended in men with non-metastatic CRPC. However, those with a rapidly rising PSA on ADT (doubling time of less than 8-10 months), are at significantly greater risk of developing metastases and death. The estimated mean survival of patients with CRPC is 9-36 months, and there is therefore an unmet need for new effective therapies.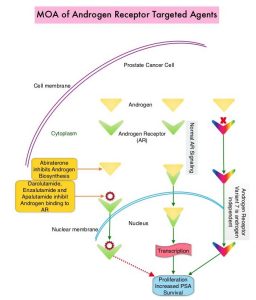
Expression of Androgen Receptor (AR) in prostate cancer is heterogeneous and this AR heterogeneity is accentuated in advanced metastatic and relapsed prostate cancer with varying degrees of AR resistance and sensitivity. Both single agent ERLEADA® (Apalutamide) and ZYTIGA® (Abiraterone acetate) in combination with Prednisone, are approved for the treatment of metastatic CRPC. They have distinct mechanisms of action on the Androgen Receptors. Apalutamide is an Androgen Receptor (AR) inhibitor that binds directly to the ligand-binding domain of the AR. Apalutamide inhibits AR nuclear translocation, inhibits DNA binding, and impedes AR-mediated transcription. Abiraterone acetate is an androgen biosynthesis inhibitor that inhibits CYP17, an enzyme expressed in testicular, adrenal, and prostatic tumor tissues, and is required for androgen biosynthesis. The ACIS trial was conducted to study the benefit of androgen annihilation by combining these two drugs (dual inhibition), for the first-line treatment of metastatic CRPC.
ACIS is a randomized, double-blind, placebo-controlled, multicenter study Phase III trial in which the efficacy and safety of single agent Apalutamide and Abiraterone acetate along with Prednisone plus ADT, was compared to placebo and Abiraterone acetate with Prednisone plus ADT, in patients with chemotherapy-naïve mCRPC. This study enrolled 982 chemo naïve patients with metastatic CRPC, who had disease progression on ADT, and were on no other life-prolonging treatment since diagnosis. Patients were randomized 1:1 to receive Apalutamide 240 mg daily along with Abiraterone acetate 1000 mg daily plus Prednisone 5 mg twice daily, all given orally (N=492) versus Placebo along with Abiraterone Acetate plus Prednisone (N=490). All patients were also on Androgen Deprivation Therapy. The median patient age was 71 years, 53% of patients had a Gleason score of 7 or more at initial diagnosis, about 85% had bone metastases, 48% had lymph node metastases and 15% had visceral metastases. Baseline characteristics were comparable in both treatment groups.
The Primary end point was radiographic Progression Free Survival (rPFS), defined from randomization date to radiographic progression date or death. Secondary end points included PSA response, Overall Survival (OS), initiation of cytotoxic chemotherapy, and pain progression.
This trial met its Primary endpoint of rPFS benefit with androgen annihilation, and the Apalutamide plus Abiraterone combination prolonged rPFS from 16.6 months to 22.6 months (HR=0.69, P<0.0001), suggesting a 31% reduction in the risk of radiographic disease progression and death. However, after 54.8 months of median follow-up, Overall Survival was numerically higher but not statistically significantly higher with Apalutamide plus Abiraterone combination (36.2 months versus 33.7 months, P=0.498). In the pre-specified subgroup analysis, the Apalutamide plus Abiraterone combination was more favorable in patients 75 years and older and in those with visceral metastases. More patients receiving Apalutamide plus Abiraterone combination had a PSA decline greater than 50%. In an exploratory analysis of biomarkers of response, tumors classified as luminal by the PAM50 signature score, or those having high AR activity expression signatures, trended towards improved rPFS and OS with the Apalutamide plus Abiraterone combination. There were no new safety signals noted with the Apalutamide plus Abiraterone combination, and based on FACT (Functional Assessment of Cancer Therapy-Prostate) -P score, Quality of Life was comparable between treatment groups.
It was concluded that the ACIS trial met its Primary endpoint, and when compared to Abiraterone with ADT, a combination of Apalutamide plus Abiraterone along with ADT demonstrated a 31% reduction in risk of radiographic progression or death, in chemo-naive mCRPC patients.
Final results from ACIS, a randomized, placebo (PBO)-controlled double-blind phase 3 study of apalutamide (APA) and abiraterone acetate plus prednisone (AAP) versus AAP in patients (pts) with chemo-naive metastatic castration-resistant prostate cancer (mCRPC). Rathkopf DE, Efstathiou E, Attard G, et al. On behalf of the ACIS investigators. J Clin Oncol 39, 2021 (suppl 6; abstr 9)
Advances with First-Line Dual Immunotherapies in Metastatic Non-Small Cell Lung Cancer
By Dr. David Waterhouse | Sponsored by Bristol Myers Squibb
Dr. Waterhouse is a paid consultant for Bristol Myers Squibb and was compensated for his role in drafting this article.
The American Cancer Society estimates that there will be nearly 229,000 new cases of lung cancer in the United States (US) alone in 2020 and nearly 136,000 lung cancer deaths.1 Historically, most patients present with metastatic disease and their long-term outlook is grim.2 However, significant progress has been made in recent years. In August 2020, Howlader et al reported that the population-level mortality from non-small cell lung cancer (NSCLC) in the US fell sharply from 2013 to 2016.3
Based on the results from Checkmate 227 Part 1a, OPDIVO, in combination with YERVOY, is indicated for the first-line treatment of adult patients with metastatic NSCLC whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.4-6 In addition, based on the results from Checkmate 9LA, OPDIVO, in combination with YERVOY and 2 cycles of platinum-doublet chemotherapy (chemo), is indicated for the first-line treatment of adult patients with metastatic or recurrent NSCLC, with no EGFR or ALK genomic tumor aberrations.4,6,7
OPDIVO and YERVOY are associated with the following Warnings and Precautions: severe and fatal immune-mediated reactions including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis with renal dysfunction, dermatologic adverse reactions, other immune-mediated adverse reactions; infusion-related reactions; complications of allogeneic hematopoietic stem cell transplantation (HSCT); embryo-fetal toxicity; and increased mortality in patients with multiple myeloma when OPDIVO is added to a thalidomide analogue and dexamethasone, which is not recommended outside of controlled clinical trials.4 Please see additional Important Safety Information for OPDIVO and YERVOY at the end of the article and US Full Prescribing Information for OPDIVO and YERVOY at https://packageinserts.bms.com/pi/pi_opdivo.pdf and https://packageinserts.bms.com/pi/pi_yervoy.pdf.
OPDIVO® (nivolumab) is a monoclonal antibody targeting programmed death receptor-1 (PD-1) that has been approved for the treatment of lung cancer.4 YERVOY® (ipilimumab) is another monoclonal antibody that works to activate the immune system by targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4).6,8
Figure 1: OPDIVO and YERVOY mechanisms of action4,6,8-14
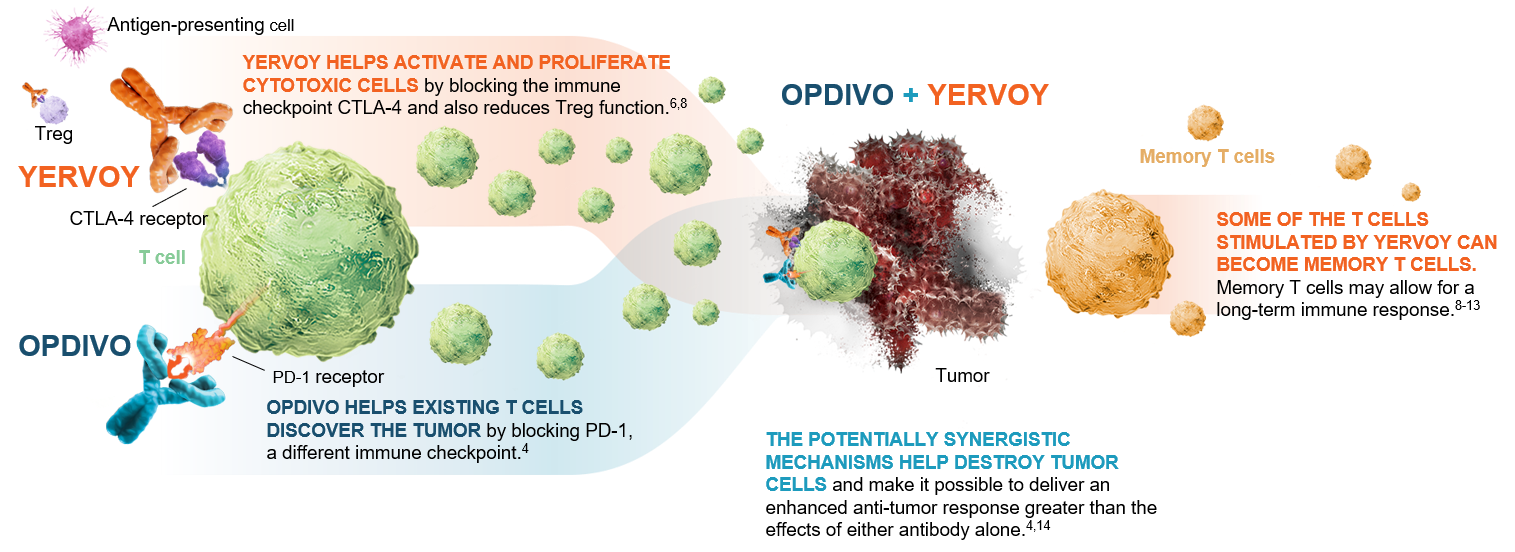 This graphic is for demonstration purposes only.
This graphic is for demonstration purposes only.
The illustrated mechanisms may vary for each patient and may not directly correlate with clinical significance.
The phase 3 Checkmate 227 and Checkmate 9LA trials investigated OPDIVO plus YERVOY-based combinations for first-line treatment of certain NSCLC patients.4 Part 1a of Checkmate 227 investigated the effects of OPDIVO + YERVOY compared with standard chemo* among patients whose tumors expressed ≥1% programmed death ligand 1 (PD-L1)4 (Figure 2).
Figure 2: Checkmate 227 Part 1a study design15
 *In Checkmate 227, patients in the comparator arm received up to 4 cycles of platinum-doublet chemo q3w; NSQ: pemetrexed + carboplatin or cisplatin, with optional pemetrexed maintenance following chemo; SQ: gemcitabine + carboplatin or cisplatin.4,16,17
*In Checkmate 227, patients in the comparator arm received up to 4 cycles of platinum-doublet chemo q3w; NSQ: pemetrexed + carboplatin or cisplatin, with optional pemetrexed maintenance following chemo; SQ: gemcitabine + carboplatin or cisplatin.4,16,17
ALK=anaplastic lymphoma kinase; DOR=duration of response; ECOG PS=Eastern Cooperative Oncology Group Performance Status; EGFR=epidermal growth factor receptor; NSQ=non-squamous; q2w=every 2 weeks; q6w=every 6 weeks; SQ=squamous.
OPDIVO + YERVOY showed a superior survival benefit compared with chemo*, with the primary analysis at a minimum follow-up of 29.3 months revealing a median overall survival (OS) of 17.1 months vs 14.9 months with chemo*, and a hazard ratio (HR) of 0.79, 95% confidence interval (CI): 0.67–0.94, P=0.0066 (Figure 3).4,16 The median progression-free survival (PFS) was 5.1 months (95% CI: 4.1–6.3) with OPDIVO + YERVOY and 5.6 months (95% CI: 4.6–5.8) with chemo* alone (HR=0.82; 95% CI: 0.69–0.97).4
The most frequent (≥2%) serious adverse reactions were pneumonia, diarrhea/colitis, pneumonitis, hepatitis, pulmonary embolism, adrenal insufficiency, and hypophysitis. Fatal adverse reactions occurred in 1.7% of patients; these included events of pneumonitis (4 patients), myocarditis, acute kidney injury, shock, hyperglycemia, multi-system organ failure, and renal failure.4 The most common (≥20%) adverse reactions were fatigue (44%), rash (34%), decreased appetite (31%), musculoskeletal pain (27%), diarrhea/colitis (26%), dyspnea (26%), cough (23%), hepatitis (21%), nausea (21%), and pruritus (21%).4 Please continue reading for more Important Safety Information for OPDIVO and YERVOY throughout.
Figure 3: Checkmate 227 OS for PD L1 ≥1% (extended 3-year follow-up analysis)4,15
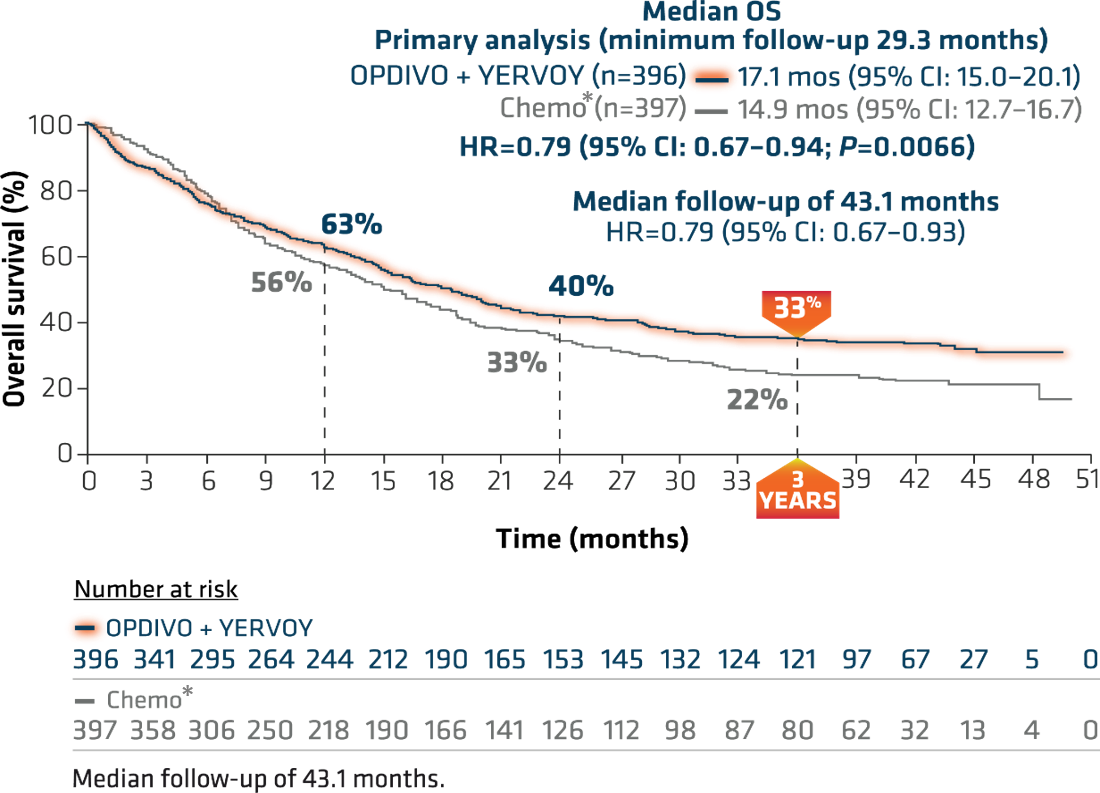
*In Checkmate 227, patients in the comparator arm received up to 4 cycles of platinum-doublet chemo q3w; NSQ: pemetrexed + carboplatin or cisplatin, with optional pemetrexed maintenance following chemo; SQ: gemcitabine + carboplatin or cisplatin.4,16,17
At the American Society for Clinical Oncology (ASCO) 2020 Annual Meeting, 3-year follow-up results from this trial were reported. With a median follow-up of more than 3 years (43.1 months), this study represents the longest median follow-up of any dual immuno-oncology (I-O)-based combination in a phase 3 clinical trial in NSCLC.15 This extended follow-up analysis showed 3-year OS rates of 33% for OPDIVO + YERVOY and 22% for chemo* (Figure 3).15
At minimum follow-up of 28.3 months, the objective response rate was 36% (95% CI: 31–41), CR=5.8%, PR=30.1% with OPDIVO + YERVOY and 30% (95% CI: 26–35), CR=1.8%, PR=28.2% with chemo*.4,16,17 The median duration of response from the extended 3-year follow-up analysis was 23.2 months (95% CI: 15.2–32.2) in patients who responded to OPDIVO + YERVOY and 6.7 months (95% CI: 5.6–7.6) with chemo* (Figure 4).15
Figure 4: Checkmate 227 DOR among responders with PD L1 ≥1% (extended 3-year follow-up analysis)15
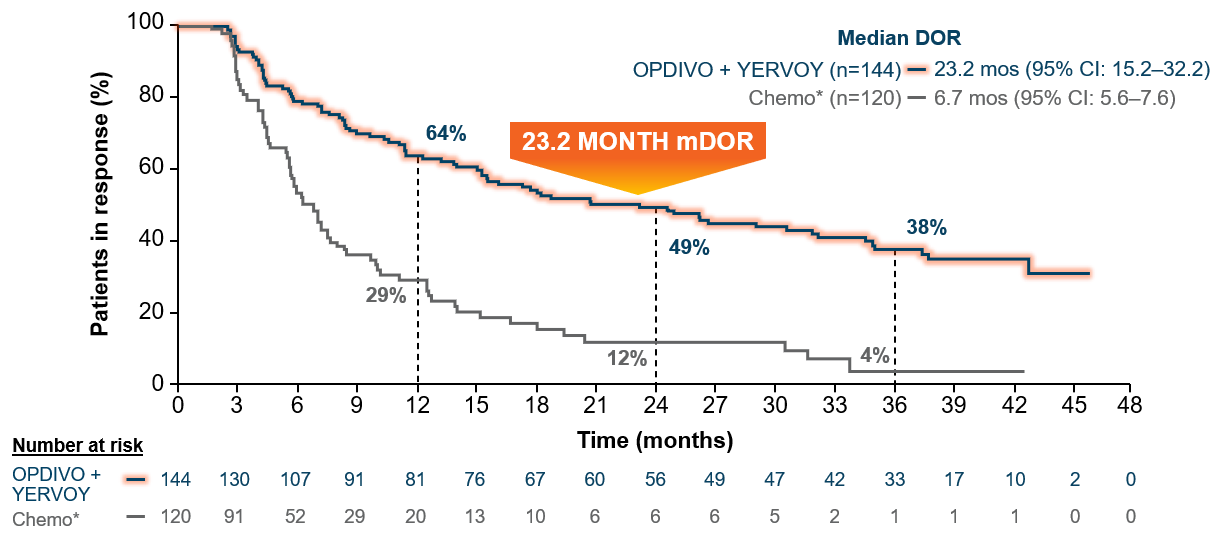
Median follow-up of 43.1 months.15
*In Checkmate 227, patients in the comparator arm received up to 4 cycles of platinum-doublet chemo q3w; NSQ: pemetrexed + carboplatin or cisplatin, with optional pemetrexed maintenance following chemo; SQ: gemcitabine + carboplatin or cisplatin.4,16,17
The 3-year data from Checkmate 227 Part 1a show the long-term durable survival of a dual immunotherapy approach.15 The FDA approved OPDIVO + YERVOY on May 15, 2020, for first-line treatment of adult patients with metastatic NSCLC whose tumors express PD-L1(≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations. With this approval, these patients with NSCLC can now be offered the option of dual I-O therapy.4,5
Also reported at ASCO 2020 were the results of Checkmate 9LA.18 Patients were randomized to receive either the combination of OPDIVO + YERVOY and 2 cycles of platinum-doublet chemo† or platinum-doublet chemo† for 4 cycles.4 This trial evaluated patients regardless of PD-L1 expression and histology (Figure 5).4
Figure 5: Checkmate 9LA study design18
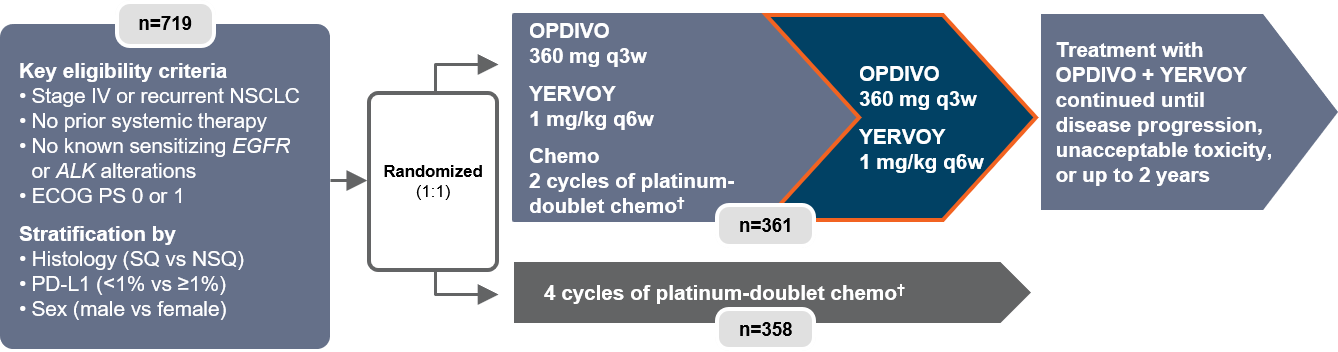
†In Checkmate 9LA, patients received 2 cycles of platinum-doublet chemo q3w in the experimental arm, and up to 4 cycles in the comparator arm; NSQ: pemetrexed + carboplatin or cisplatin (optional pemetrexed maintenance therapy in comparator arm only); SQ: paclitaxel + carboplatin.4
q3w=every three weeks.
The trial showed a superior benefit in OS for patients treated with OPDIVO + YERVOY with limited chemo† compared to those who received chemo† alone.18 At the pre-specified interim analysis at 8.1 months, the median OS was 14.1 months vs 10.7 months (HR=0.69, 96.71% CI: 0.55-0.87, P=0.0006).4 Median PFS per blinded independent central review (BICR) at minimum follow-up of 6.5 months was 6.8 months among patients who received OPDIVO + YERVOY with chemo†, and 5.0 months among patients receiving chemo† (HR=0.70, 97.48% CI: 0.57-0.86).4 Confirmed ORR per BICR at minimum follow-up of 6.5 months was 38% (95% CI: 33-43) and 25% (95% CI: 21-30) respectively.4,18
A follow-up analysis performed at 12.7 months showed median OS of 15.6 months with OPDIVO + YERVOY with chemo† and 10.9 months with chemo† alone with HR of 0.66 (95% CI: 0.55-0.80) (Figure 6).4,18 OS was consistent across PD-L1 expression levels at minimum follow-up of 8.1 months, with median OS of 14.0 months (95% CI:13.2-NR) and 10.0 months (95% CI: 7.7-13.7) in patients treated with OPDIVO + YERVOY with limited chemo† and chemo† respectively in the PD-L1 <1% sub-population (HR=0.65), and median OS of 14.2 months (95% CI:13.1-NR) and 10.6 months (95% CI: 9.4-12.6) respectively (HR=0.67) in the PD-L1 ≥1% sub-population.19
Figure 6: Checkmate 9LA overall survival (extended follow-up)18
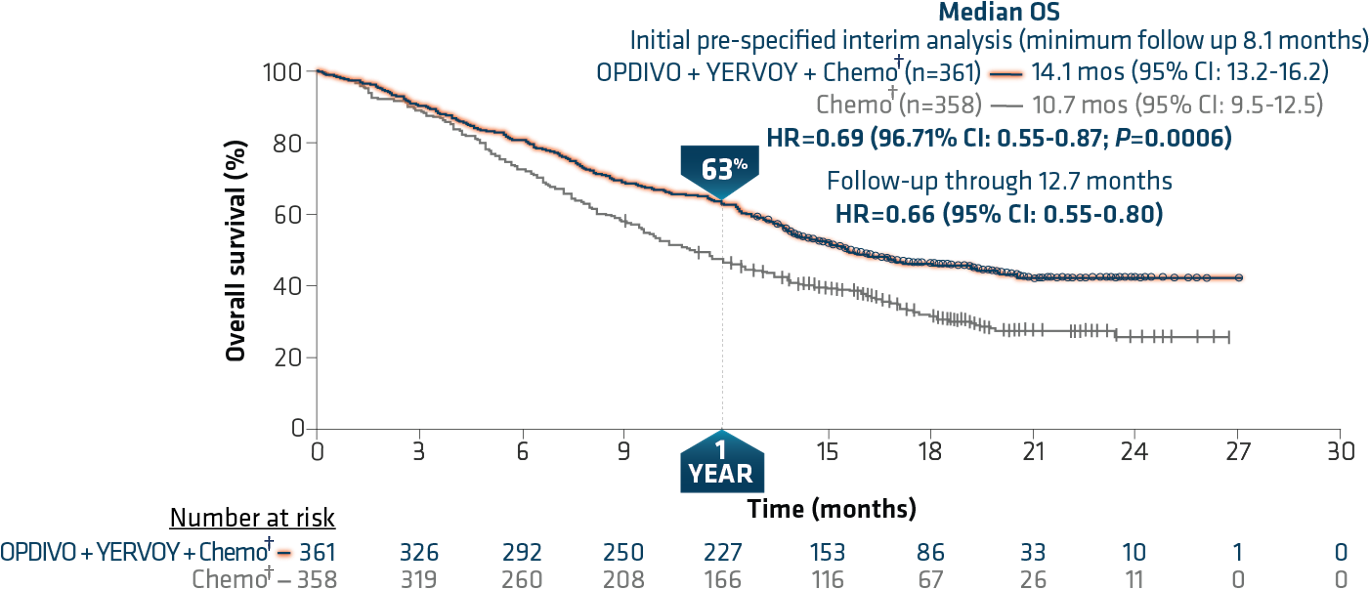
Minimum follow-up of 12.7 months.
†In Checkmate 9LA, patients received 2 cycles of platinum-doublet chemo q3w in the experimental arm, and up to 4 cycles in the comparator arm; NSQ: pemetrexed + carboplatin or cisplatin (optional pemetrexed maintenance therapy in comparator arm only); SQ: paclitaxel + carboplatin.4
In this study, the most frequent (>2%) serious adverse reactions were pneumonia, diarrhea, febrile neutropenia, anemia, acute kidney injury, musculoskeletal pain, dyspnea, pneumonitis, and respiratory failure. Fatal adverse reactions occurred in 7 (2%) patients, and included hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia.4 The most common (>20%) adverse reactions were fatigue (49%), musculoskeletal pain (39%), nausea (32%), diarrhea (31%), rash (30%), decreased appetite (28%), constipation (21%), and pruritus (21%).4 Please continue reading for more Important Safety Information for OPDIVO and YERVOY throughout. The FDA approved OPDIVO, in combination with YERVOY and 2 cycles of platinum-doublet chemo, for the first-line treatment of adult patients with metastatic or recurrent NSCLC with no EGFR or ALK genomic tumor aberrations in May 2020.4,7
With the approval of both Checkmate 227 and Checkmate 9LA regimens as first-line therapies, I am pleased to be able to offer metastatic NSCLC patients with additional options. Checkmate 227 provides appropriate mNSCLC patients with a chemo-free, dual I-O option with long-term, durable survival. Additionally, the Checkmate 9LA regimen with dual I-O plus limited chemo† has shown superior OS, and consistent OS, regardless of PD-L1 expression in recurrent/metastatic NSCLC patients.4,18
*In Checkmate 227, patients in the comparator arm received up to 4 cycles of platinum-doublet chemo q3w; NSQ: pemetrexed + carboplatin or cisplatin, with optional pemetrexed maintenance following chemo; SQ: gemcitabine + carboplatin or cisplatin.4,16,17
†In Checkmate 9LA, patients received 2 cycles of platinum-doublet chemo q3w in the experimental arm, and up to 4 cycles in the comparator arm; NSQ: pemetrexed + carboplatin or cisplatin (optional pemetrexed maintenance therapy in comparator arm only); SQ: paclitaxel + carboplatin.4
IMPORTANT SAFETY INFORMATION
Severe and Fatal Immune-Mediated Adverse Reactions
Immune-mediated adverse reactions listed herein may not include all possible severe and fatal immune-mediated adverse reactions.
Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue. While immune-mediated adverse reactions usually manifest during treatment, they can also occur after discontinuation of OPDIVO or YERVOY. Early identification and management are essential to ensure safe use of OPDIVO and YERVOY. Monitor for signs and symptoms that may be clinical manifestations of underlying immune-mediated adverse reactions. Evaluate clinical chemistries including liver enzymes, creatinine, adrenocorticotropic hormone (ACTH) level, and thyroid function at baseline and periodically during treatment with OPDIVO and before each dose of YERVOY. In cases of suspected immune-mediated adverse reactions, initiate appropriate workup to exclude alternative etiologies, including infection. Institute medical management promptly, including specialty consultation as appropriate.
Withhold or permanently discontinue OPDIVO and YERVOY depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information). In general, if OPDIVO or YERVOY interruption or discontinuation is required, administer systemic corticosteroid therapy (1 to 2 mg/kg/day prednisone or equivalent) until improvement to Grade 1 or less. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Consider administration of other systemic immunosuppressants in patients whose immune-mediated adverse reactions are not controlled with corticosteroid therapy. Toxicity management guidelines for adverse reactions that do not necessarily require systemic steroids (e.g., endocrinopathies and dermatologic reactions) are discussed below.
Immune-Mediated Pneumonitis
OPDIVO and YERVOY can cause immune-mediated pneumonitis. The incidence of pneumonitis is higher in patients who have received prior thoracic radiation. In NSCLC patients receiving OPDIVO 3 mg/kg every 2 weeks with YERVOY 1 mg/kg every 6 weeks, immune-mediated pneumonitis occurred in 9% (50/576) of patients, including Grade 4 (0.5%), Grade 3 (3.5%), and Grade 2 (4.0%). Four patients (0.7%) died due to pneumonitis.
Immune-Mediated Colitis
OPDIVO and YERVOY can cause immune-mediated colitis, which may be fatal. A common symptom included in the definition of colitis was diarrhea. Cytomegalovirus (CMV) infection/reactivation has been reported in patients with corticosteroid-refractory immune-mediated colitis. In cases of corticosteroid-refractory colitis, consider repeating infectious workup to exclude alternative etiologies.
Immune-Mediated Hepatitis
OPDIVO and YERVOY can cause immune-mediated hepatitis.
Immune-Mediated Endocrinopathies
OPDIVO and YERVOY can cause primary or secondary adrenal insufficiency, immune-mediated hypophysitis, immune-mediated thyroid disorders, and Type 1 diabetes mellitus, which can present with diabetic ketoacidosis. Withhold OPDIVO and YERVOY depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information). For Grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically indicated. Hypophysitis can present with acute symptoms associated with mass effect such as headache, photophobia, or visual field defects. Hypophysitis can cause hypopituitarism; initiate hormone replacement as clinically indicated. Thyroiditis can present with or without endocrinopathy. Hypothyroidism can follow hyperthyroidism; initiate hormone replacement or medical management as clinically indicated. Monitor patients for hyperglycemia or other signs and symptoms of diabetes; initiate treatment with insulin as clinically indicated.
Immune-Mediated Nephritis with Renal Dysfunction
OPDIVO and YERVOY can cause immune-mediated nephritis.
Immune-Mediated Dermatologic Adverse Reactions
OPDIVO can cause immune-mediated rash or dermatitis. Exfoliative dermatitis, including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug rash with eosinophilia and systemic symptoms (DRESS) has occurred with PD-1/PD-L1 blocking antibodies. Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate nonexfoliative rashes.
YERVOY can cause immune-mediated rash or dermatitis, including bullous and exfoliative dermatitis, SJS, TEN, and DRESS. Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate non-bullous/exfoliative rashes.
Withhold or permanently discontinue OPDIVO and YERVOY depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information).
Other Immune-Mediated Adverse Reactions
The following clinically significant immune-mediated adverse reactions occurred at an incidence of <1% (unless otherwise noted) in patients who received OPDIVO monotherapy or OPDIVO in combination with YERVOY or were reported with the use of other PD-1/PD-L1 blocking antibodies. Severe or fatal cases have been reported for some of these adverse reactions: cardiac/vascular: myocarditis, pericarditis, vasculitis; nervous system: meningitis, encephalitis, myelitis and demyelination, myasthenic syndrome/myasthenia gravis (including exacerbation), Guillain-Barré syndrome, nerve paresis, autoimmune neuropathy; ocular: uveitis, iritis, and other ocular inflammatory toxicities can occur; gastrointestinal: pancreatitis to include increases in serum amylase and lipase levels, gastritis, duodenitis; musculoskeletal and connective tissue: myositis/polymyositis, rhabdomyolysis, and associated sequelae including renal failure, arthritis, polymyalgia rheumatica; endocrine: hypoparathyroidism; other (hematologic/immune): hemolytic anemia, aplastic anemia, hemophagocytic lymphohistiocytosis (HLH), systemic inflammatory response syndrome, histiocytic necrotizing lymphadenitis (Kikuchi lymphadenitis), sarcoidosis, immune thrombocytopenic purpura, solid organ transplant rejection.
In addition to the immune-mediated adverse reactions listed above, across clinical trials of YERVOY monotherapy or in combination with OPDIVO, the following clinically significant immune-mediated adverse reactions, some with fatal outcome, occurred in <1% of patients unless otherwise specified: nervous system: autoimmune neuropathy (2%), myasthenic syndrome/myasthenia gravis, motor dysfunction; cardiovascular: angiopathy, temporal arteritis; ocular: blepharitis, episcleritis, orbital myositis, scleritis; gastrointestinal: pancreatitis (1.3%); other (hematologic/immune): conjunctivitis, cytopenias (2.5%), eosinophilia (2.1%), erythema multiforme, hypersensitivity vasculitis, neurosensory hypoacusis, psoriasis.
Some ocular IMAR cases can be associated with retinal detachment. Various grades of visual impairment, including blindness, can occur. If uveitis occurs in combination with other immune-mediated adverse reactions, consider a Vogt-Koyanagi-Harada–like syndrome, which has been observed in patients receiving YERVOY, as this may require treatment with systemic corticosteroids to reduce the risk of permanent vision loss.
Infusion-Related Reactions
OPDIVO and YERVOY can cause severe infusion-related reactions. Discontinue OPDIVO and YERVOY in patients with severe (Grade 3) or life-threatening (Grade 4) infusion-related reactions. Interrupt or slow the rate of infusion in patients with mild (Grade 1) or moderate (Grade 2) infusion-related reactions.
Complications of Allogeneic Hematopoietic Stem Cell Transplantation
Fatal and other serious complications can occur in patients who receive allogeneic hematopoietic stem cell transplantation (HSCT) before or after being treated with OPDIVO or YERVOY. Transplant-related complications include hyperacute graft-versus-host-disease (GVHD), acute GVHD, chronic GVHD, hepatic veno-occlusive disease (VOD) after reduced intensity conditioning, and steroid-requiring febrile syndrome (without an identified infectious cause). These complications may occur despite intervening therapy between OPDIVO or YERVOY and allogeneic HSCT.
Follow patients closely for evidence of transplant-related complications and intervene promptly. Consider the benefit versus risks of treatment with OPDIVO and YERVOY prior to or after an allogeneic HSCT.
Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal studies, OPDIVO and YERVOY can cause fetal harm when administered to a pregnant woman. The effects of YERVOY are likely to be greater during the second and third trimesters of pregnancy. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with OPDIVO and YERVOY and for at least 5 months after the last dose.
Increased Mortality in Patients with Multiple Myeloma when OPDIVO is Added to a Thalidomide Analogue and Dexamethasone
In randomized clinical trials in patients with multiple myeloma, the addition of OPDIVO to a thalidomide analogue plus dexamethasone resulted in increased mortality. Treatment of patients with multiple myeloma with a PD-1 or PD-L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled clinical trials.
Lactation
There are no data on the presence of OPDIVO or YERVOY in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment and for 5 months after the last dose.
Serious Adverse Reactions
In Checkmate 227, serious adverse reactions occurred in 58% of patients (n=576). The most frequent (≥2%) serious adverse reactions were pneumonia, diarrhea/colitis, pneumonitis, hepatitis, pulmonary embolism, adrenal insufficiency, and hypophysitis. Fatal adverse reactions occurred in 1.7% of patients; these included events of pneumonitis (4 patients), myocarditis, acute kidney injury, shock, hyperglycemia, multi-system organ failure, and renal failure. In Checkmate 9LA, serious adverse reactions occurred in 57% of patients (n=358). The most frequent (>2%) serious adverse reactions were pneumonia, diarrhea, febrile neutropenia, anemia, acute kidney injury, musculoskeletal pain, dyspnea, pneumonitis, and respiratory failure. Fatal adverse reactions occurred in 7 (2%) patients, and included hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia.
Common Adverse Reactions
In Checkmate 227, the most common (≥20%) adverse reactions were fatigue (44%), rash (34%), decreased appetite (31%), musculoskeletal pain (27%), diarrhea/colitis (26%), dyspnea (26%), cough (23%), hepatitis (21%), nausea (21%), and pruritus (21%). In Checkmate 9LA, the most common (>20%) adverse reactions were fatigue (49%), musculoskeletal pain (39%), nausea (32%), diarrhea (31%), rash (30%), decreased appetite (28%), constipation (21%), and pruritus (21%).
Please see U.S. Full Prescribing Information for OPDIVO and YERVOY:
• https://packageinserts.bms.com/pi/pi_opdivo.pdf
• https://packageinserts.bms.com/pi/pi_yervoy.pdf
References:
1. Key statistics for lung cancer. American Cancer Society. Reviewed October 1, 2019. Revised January 8, 2020. Accessed October 7, 2020. https://www.cancer.org/cancer/lung-cancer/about/key-statistics.html.
2. Lung and bronchus cancer – cancer stat facts. National Cancer Institute. Accessed October 7, 2020. https://seer.cancer.gov/statfacts/html/lungb.html.
3. Howlader N, Forjaz G, Mooradian MJ, et al. The effect of advances in lung-cancer treatment on population mortality. N Engl J Med. 2020;383:640-649.
4. OPDIVO [package insert]. Princeton, NJ: Bristol-Myers Squibb Company.
5. FDA approval for Checkmate 227. Accessed October 12, 2020. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-nivolumab-plus-ipilimumab-first-line-mnsclc-pd-l1-tumor-expression-1.
6. YERVOY [package insert]. Princeton, NJ: Bristol-Myers Squibb Company.
7. FDA approval for Checkmate 9LA. Accessed October 12, 2020. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-nivolumab-plus-ipilimumab-and-chemotherapy-first-line-treatment-metastatic-nsclc.
8. Weber JS, Hamid O, Chasalow SD, et al. Ipilimumab increases activated T cells and enhances humoral immunity in patients with advanced melanoma. J Immunother. 2012;35:89-97.
9. Farber DL, Yudanin NA, and Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014;14(1):24-35.
10. Ansell SM, Hurvitz SA, Koenig PA, et al. Phase I study of ipilimumab, an anti–CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non–Hodgkin lymphoma. Clin Cancer Res. 2009;15(20):6446-6453.
11. Felix J, Lambert J, Roelens M, et al. Ipilimumab reshapes T cell memory subsets in melanoma patients with clinical response. Oncoimmunology. 2016;5(7):e1136045.
12. Pedicord VA, Montalvo W, Leiner IM, and Allison JP. Single dose of anti–CTLA-4 enhances CD8+ T-cell memory formation, function, and maintenance. Proc Natl Acad Sci USA. 2011;108(1):266-271.
13. de Coaña YP, Wolodarski M, Poschke I, et al. Ipilimumab treatment decreases monocytic MDSCs and increases CD8 effector memory T cells in long-term survivors with advanced melanoma. Oncotarget. 2017;8(13):21539-21553.
14. Buchbinder EI and Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39:98-106.
15. Ramalingam S, Ciuleanu T-E, Pluzanski A, et al. Nivolumab + ipilimumab versus platinum-doublet chemotherapy as first-line treatment for advanced non-small cell lung cancer: Three-year update from Checkmate 227 Part 1. Oral presentation at ASCO 2020. Abstract 9500.
16. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N Engl J Med. 2019;381:2020-2031.
17. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N Engl J Med. 2019;381:2020-2031. [supplementary appendix].
18. Reck M, Ciuleanu T-E, Cobo M, et al. Nivolumab + ipilimumab + 2 cycles of platinum-doublet chemotherapy vs 4 cycles chemotherapy as first-line treatment for stage IV/recurrent NSCLC: Checkmate 9LA. Oral presentation at ASCO 2020. Abstract 9501.
19. Data on file. NIVO 566. Princeton, NJ: Bristol-Myers Squibb Company.