SUMMARY: The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the immune system T cells. Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152) and PD-1(Programmed cell Death 1). Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response. OPDIVO® is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4. In the CheckMate-227, Part 1, Phase III trial, a combination of OPDIVO® plus YERVOY®, significantly improved Overall Survival (OS), Progression Free Survival (PFS), Objective Response Rates (ORR) and Duration of Response, compared to chemotherapy, independent of PD-L1 expression level. The authors in this study hypothesized that a limited course of chemotherapy combined with OPDIVO® plus YERVOY® could provide rapid disease control, while building on the durable Overall Survival benefit seen with dual PD-1 and CTLA-4 inhibition, as well as minimizing the toxicities associated with a full course of chemotherapy.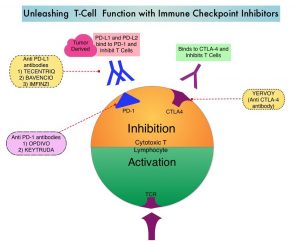
CheckMate-9LA is a randomized, open-label, multi-center, Phase III trial which evaluated the benefit of a combination of OPDIVO® plus YERVOY®, and 2 cycles of Platinum-doublet chemotherapy (experimental arm) versus Platinum-doublet chemotherapy (control arm) for 4 cycles, followed by optional Pemetrexed maintenance therapy, as a first-line treatment in patients with metastatic or recurrent NSCLC, regardless of PD-L1 status and histology. In this study, 719 adults treatment naïve patients with histologically confirmed Stage IV/recurrent NSCLC, with ECOG Performance Status 0-1, and no known sensitizing EGFR/ALK alterations, were randomly assigned 1:1 to receive OPDIVO® 360 mg every 3 weeks plus YERVOY® 1 mg/kg every 6 weeks and 2 cycles of platinum-doublet chemotherapy (N=361), or 4 cycles of platinum-doublet chemotherapy alone (N=358). Chemotherapy was based on histology. Patients with non-squamous NSCLC in the chemo-only randomized group could receive optional Pemetrexed maintenance treatment. Patients were treated with immunotherapy until disease progression, unacceptable toxicity, or for 2 years. Patients were stratified by PD-L1 status (less than 1% versus 1% or more), sex, and histology (squamous versus non-squamous). Demographics in treatment groups were well balanced. Crossover between treatment groups was not permitted. However, at physician discretion, patients could receive subsequent immunotherapy upon discontinuation of study treatment in either group.
The Primary end point was Overall Survival (OS). Secondary endpoints included Progression Free Survival (PFS), Objective Response Rate (ORR) and efficacy by PD-L1 subgroups. PFS2 was a pre-specified exploratory endpoint and was defined as time from randomization, to objectively documented progression after the next line of therapy, or to death from any cause, whichever occurred first. At a preplanned interim analysis after a minimum follow up 8.1 months, this trial met its primary and secondary endpoints, showing statistically significant improvements in OS, PFS, and Objective Response Rate (ORR), when compared to chemotherapy alone. This clinical benefit was noted across tumor PD-L1 expression levels and histologies.
The authors in this publication reported updated efficacy and safety outcomes, along with Progression-Free Survival (PFS) after next line of treatment (PFS2), Treatment-Related Adverse Events (TRAEs) by treatment cycle, and efficacy outcomes in patients who discontinued all treatment components in the experimental treatment group due to TRAEs, from the CheckMate 9LA Phase III trial. The minimum follow up for OS was 24.4 months. The majority of patients (93%) received two cycles of chemotherapy and 13% completed the maximum 2 years of immunotherapy treatment. The median number of doses was 9.0 for OPDIVO® and 4.0 for YERVOY®. In the control arm, 75% of patients received four cycles of chemotherapy and 67% patients who had non-squamous tumor histology receiving Pemetrexed maintenance. About 29% patients in the control arm had completed the full four cycles of chemotherapy without optional Pemetrexed maintenance therapy. The median duration of therapy was 6.1 months in the experimental arm and 2.5 months in the control arm.
With a median follow up of 30.7 months, OPDIVO® plus YERVOY® with a limited course of chemotherapy continued to prolong Overall Survival (OS), when compared to chemotherapy (Median OS 15.8 versus 11.0 months; HR=0.72). The 2-year OS rate was 38% versus 26%. This OS benefit was observed across most key subgroups including those with PD-L1 expression of less than 1%, more than 1%, as well as by histology. More importantly, patients with pretreated CNS metastases at baseline had a median OS of 19.9 months in the experimental group versus 7.9 months in the control group, respectively (HR=0.47).
PFS continued to be prolonged in the experimental group compared to the control group, with an Hazard Ratio of 0.67 and 2-year PFS rates of 20% versus 8%, respectively. The ORR was 38% in the experimental group and 25% in the control group (P=0.0003). 34% versus 12% of all responses respectively, were ongoing at 2 years. The median PFS2 in all randomized patients was 13.9 months in the experimental group and 8.7 months in the control group (HR=0.66). Again, PFS2 also favored the experimental arm over the control arm in subgroups by PD-L1 expression, and by histology.
No new safety signals were observed and majority of Grade 3/4 toxicities were mostly observed during the first two treatment cycles in the experimental group. In patients who discontinued all components of the experimental treatment (OPDIVO® plus YERVOY® with chemotherapy) due to toxicities (N=61), the median OS was 27.5 months and 56% of responders had an ongoing response, more than 1 year after discontinuation of therapy. After discontinuing the experimental regimen, patients remained treatment-free for a median of 11.9 months and had a 48% chance of being treatment-free at 1 year.
The researchers concluded that with a 2-year minimum follow-up, OPDIVO® plus YERVOY® with two cycles of chemotherapy provided durable efficacy benefits over conventional chemotherapy, with a manageable safety profile. They added that this treatment regimen remains an efficacious first line treatment of advanced Non Small Cell Lung Cancer.
First-line nivolumab plus ipilimumab with two cycles of chemotherapy versus chemotherapy alone (four cycles) in advanced non-small-cell lung cancer: CheckMate 9LA 2-year update. Reck M, Ciuleanu T-E, Cobo M, et al. https://doi.org/10.1016/j.esmoop.2021.100273