
The FDA on October 7, 2021, granted recognition to a partial listing of the Memorial Sloan Kettering Cancer Center’s Oncology Knowledge Base (OncoKB) as the first tumor mutation database to be included in the Public Human Genetic Variant Databases.
Author: RR
TECARTUS® (Brexucabtagene autoleucel)
The FDA on October 1, 2021, approved TECARTUS® (Brexucabtagene autoleucel) for adult patients with Relapsed or Refractory B-cell precursor Acute Lymphoblastic Leukemia (ALL). TECARTUS® is a product of Kite Pharma, Inc.
JAKAFI® (Ruxolitinib)
The FDA on September 22, 2021 approved JAKAFI® (Ruxolitinib) for chronic Graft-Versus-Host Disease (cGVHD) after failure of one or two lines of systemic therapy in adult and pediatric patients 12 years and older. JAKAFI® is a product of Incyte Corp.
TIVDAK® (Tisotumab vedotin-tftv)
The FDA on September 20, 2021 granted accelerated approval to TIVDAK®, a tissue factor-directed antibody and microtubule inhibitor conjugate, for adult patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy.
TIVDAK® is a product of Seagen Inc.
FDA Approves PSMA Targeted Therapy for Metastatic Castrate Resistant Prostate Cancer
SUMMARY: The FDA on March 23, 2022, approved PLUVICTO® (Lutetium Lu 177 vipivotide tetraxetan) for the treatment of adult patients with Prostate-Specific Membrane Antigen (PSMA)-positive metastatic Castration-Resistant Prostate Cancer (mCRPC), who had been treated with Androgen Receptor (AR) pathway inhibition and Taxane-based chemotherapy. The FDA also approved LOCAMETZ® (Gallium Ga 68 gozetotide), a radioactive diagnostic agent for Positron Emission Tomography (PET) of PSMA-positive lesions, including selection of patients with metastatic prostate cancer for whom PLUVICTO® PSMA-directed therapy is indicated. LOCAMETZ® is the first radioactive diagnostic agent approved for patient selection in the use of a radioligand therapeutic agent.
The development and progression of prostate cancer is driven by androgens. Androgen Deprivation Therapy (ADT) or testosterone suppression has therefore been the cornerstone of treatment of advanced prostate cancer, and is the first treatment intervention. Approximately 10-20% of patients with advanced Prostate cancer will progress to Castration Resistant Prostate Cancer (CRPC) within five years during ADT, and over 80% of these patients will have metastatic disease at the time of CRPC diagnosis. Among those patients without metastases at CRPC diagnosis, 33% are likely to develop metastases within two years. Progression to Castration Resistant Prostate Cancer (CRPC) often manifests itself with a rising PSA (Prostate Specific Antigen) and the estimated mean survival of patients with CRPC is 9-36 months, and there is therefore an unmet need for new effective therapies.
Prostate-Specific Membrane Antigen (PSMA) is a Type II cell membrane glycoprotein that is selectively expressed in prostate cells, with high levels of expression in prostatic adenocarcinoma. PSMA is a therefore an excellent target for molecular imaging and therapeutics, due to its high specificity for prostate cancer.
PLUVICTO® is a radiopharmaceutical that targets PSMA. It is comprised of Lutetium-177, a cytotoxic radionuclide , linked to the ligand PSMA-617, a small molecule designed to bind with high affinity to PSMA. Radioligand therapy with PLUVICTO® targets PSMA and releases its payload of lethal beta radiation into the prostate cancer cell. The antitumor activity and safety PLUVICTO® have been established previously in a Phase II study (Lancet Oncol. 2018;19:825-833).
VISION is an international, randomized, open-label Phase III study in which the benefit of PLUVICTO® was evaluated in men with PSMA-positive mCRPC, previously treated with second generation Androgen Receptor signaling pathway inhibitor (XTANDI® -Enzalutamide or ZYTIGA®-Abiraterone acetate), and 1 or 2 taxane chemotherapy regimens. In this trial, 831 patients were randomized 2:1 to receive PLUVICTO® 7.4 GBq (200 mCi) every 6 weeks for up to a total of 6 cycles plus Standard of Care as determined by the treating physician (N=551), or Standard of Care only (N=280). Both treatment groups were well balanced and this trial excluded patients treated with XOFIGO® (Radium-223). All enrolled patients received a GnRH analog or had prior bilateral orchiectomy and had a castrate level or serum/plasma testosterone of lower than 50ng/dL. PET imaging with LOCAMETZ® was used to determine PSMA positivity by central review. PSMA-positive mCRPC was defined as having at least one tumor lesion with LOCAMETZ® uptake greater than normal liver. Patients were excluded from enrollment if any lesions exceeding certain size criteria in the short axis had uptake less than or equal to uptake in normal liver. The Primary endpoints were radiographic Progression Free Survival (rPFS) by Independent Central Review (ICR) and Overall Survival (OS). Secondary endpoints included Objective Response Rate (ORR), Disease Control Rate (DCR), and time to first Symptomatic Skeletal Event (SSE). The median study follow up was 20.9 months.
Treatment with the combination of PLUVICTO® plus Standard of Care resulted in a statistically significant improvement in the Primary endpoints of Overall Survival and radiographic Progression Free Survival. PLUVICTO® plus Standard of Care significantly improved rPFS by 60%, compared to Standard of Care alone (median rPFS 8.7 versus 3.4 months, HR=0.40; P<0.001). The median OS was also significantly improved by 38% in the study group (median OS 15.3 versus 11.3 months, HR=0.62; P<0.001). All key secondary endpoints including Objective Response Rate, Disease Control Rate, and time to first Symptomatic Skeletal Event were statistically significant, and in favor of PLUVICTO® plus Standard of Care. The most common adverse reactions in patients receiving PLUVICTO® were fatigue, dry mouth, nausea, decreased appetite, constipation, anemia, lymphopenia, thrombocytopenia, hypocalcemia and hyponatremia.
It was concluded that radioligand therapy with PLUVICTO® significantly improved radiographic Progression Free Survival and Overall Survival when added to Standard of Care, compared with Standard of Care alone, in men with PSMA-positive metastatic Castration Resistant Prostate Cancer.
Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. Sartor O, de Bono J, Chi KN, et al. N Engl J Med 2021; 385:1091-1103.
FDA Approves Single Agent KEYTRUDA® for Advanced Endometrial Carcinoma
SUMMARY: The FDA on March 21, 2022 approved KEYTRUDA® (Pembrolizumab) as a single agent, for patients with advanced endometrial carcinoma that is MicroSatellite Instability-High (MSI-H) or MisMatch Repair deficient (dMMR), as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting, and who are not candidates for curative surgery or radiation. The FDA also approved VENTANA MMR RxDx Panel (Ventana Medical Systems/Roche Tissue Diagnostics) as a companion diagnostic device to select patients with dMMR in solid tumors that are eligible for treatment with KEYTRUDA® The FDA previously approved the FoundationOne CDx (F1CDx, Foundation Medicine, Inc.) as a companion diagnostic device to select patients with MSI-H in solid tumors that are eligible for treatment with KEYTRUDA®.
The American Cancer Society estimates that approximately 65,950 new cases of uterine cancer will be diagnosed in 2022 and about 12,550 individuals will die of the disease. Endometrial carcinoma is the second most prevalent gynecologic cancer in women worldwide, and its incidence has been increasing. Risk factors include age, factors that influence hormone levels such as obesity and estrogen replacement therapy, family history, diet and exercise, drugs such as Tamoxifen, etc. Patients with advanced or recurrent endometrial cancer are often treated with a combination of Carboplatin and Paclitaxel. Treatment options following failure of first-line therapy for this patient group however are limited, with single agent response rates of 10-15% and 5-year survival rates of approximately 17%.
The DNA MisMatchRepair (MMR) system is responsible for molecular surveillance and works as an editing tool that identifies errors within the microsatellite regions of DNA and removes them. Defective MMR system leads to MSI (Micro Satellite Instability) and hypermutation, with the expression of tumor-specific neoantigens at the surface of cancer cells, triggering an increase in CD3-positive, CD8-positive, and Programmed Death-1 (PD-1) expressing Tumor Infiltrating Lymphocytes and Programmed Death Ligand-1 (PD-L1) expressing intraepithelial and peritumoral immune cells, compared with MicroSatellite Stable cancers. This results in an enhanced antitumor immune response.
MSI is therefore a hallmark of defective/deficient DNA MisMatchRepair (dMMR) system. Defective MMR can be a sporadic or heritable event and can manifest as a germline mutation occurring in MMR genes including MLH1, MSH2, MSH6 and PMS2. This produces Lynch Syndrome often called Hereditary Nonpolyposis Colorectal Carcinoma-HNPCC, an Autosomal Dominant disorder that is often associated with a high risk for Colorectal and Endometrial carcinoma, as well as several other malignancies including Ovary, Stomach, Small bowel, Hepatobiliary tract, Brain and Skin. MSI is a hallmark of Lynch Syndrome-associated cancers. MSI high tumors tend to have better outcomes and this has been attributed to the abundance of Tumor Infiltrating Lymphocytes in these tumors from increase immunogenicity. These tumors therefore are susceptible to blockade with Immune Checkpoint Inhibitors.
MSI testing is performed using a PCR or NGS based assay and MSI-High refers to instability at 2 or more of the 5 mononucleotide repeat markers and MSI-Low refers to instability at 1 of the 5 markers. Patients are considered Micro Satellite Stable (MSS) if no instability occurs. MSI-L and MSS are grouped together because MSI-L tumors are uncommon and behave similar to MSS tumors. Tumors considered MSI-H have deficiency of one or more of the DNA MMR genes. MMR gene deficiency can be detected by ImmunoHistoChemistry (IHC). NCCN Guidelines recommend MMR or MSI testing for all patients with a history of Colon or Rectal cancer. Unlike Colorectal and Endometrial cancer, where MSI-H/dMMR testing is routinely undertaken, the characterization of Lynch Syndrome across heterogeneous MSI-H/dMMR tumors is unknown.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells. The FDA in 2017 granted accelerated approval to KEYTRUDA® for patients with advanced MSI-High or dMMR solid tumors, that have progressed following prior treatment, and who have no satisfactory alternative treatment options. This has led to routine MSI-H/dMMR testing in advanced solid tumors. The FDA in 2021 also approved KEYTRUDA® in combination with the multireceptor Tyrosine Kinase Inhibitor LENVIMA® (Lenvatinib) for patients with advanced endometrial carcinoma, irrespective of tumor MSI status based on the KEYNOTE-146 study.
KEYNOTE-158 is a multicenter, nonrandomized, open-label, multicohort, Phase II trial of KEYTRUDA® evaluating predictive biomarkers, in patients with advanced unresectable and/or metastatic solid tumors, who had progressed on standard of care therapy. The present FDA approval was based on the results from a total of 90 patients with MSI-H/dMMR endometrial cancer, who were enrolled in cohort D (11 patients) and cohort K (79 patients) of KEYNOTE-158 trial. This group of previously treated patients received KEYTRUDA® 200 mg IV once every 3 weeks for 35 cycles. The median patient age was 64 years, 48% had received 2 or more lines of prior therapy, and the majority of patients (68%) had received prior radiation therapy. The median duration of treatment was 8.3 months. The Primary end point was Objective Response Rate (ORR) by independent central radiologic review. Secondary end points included Duration of Response, Progression Free Survival (PFS), Overall Survival (OS), and Safety.
The Objective Response Rate was 48%, and median Duration of Response was not reached after a median follow up of 42.6 months. The median PFS was 13.1 months, and median Overall Survival was Not Reached. No new safety signals were identified and the immune-mediated adverse events or infusion reactions occurred in 28% of patients and 7% were Grades 3-4, with no fatal events.
It was concluded that KEYTRUDA® demonstrated robust and durable antitumor activity with manageable toxicity in patients with advanced MSI-H/dMMR endometrial cancer, and should be considered as a treatment option for patients with advanced MSI-H/dMMR endometrial cancer, following failure on prior therapy.
Pembrolizumab in Patients With Microsatellite Instability–High Advanced Endometrial Cancer: Results From the KEYNOTE-158 Study. O’Malley DM, Bariani GM, Cassier PA, et al. DOI: 10.1200/JCO.21.01874 Journal of Clinical Oncology 40, no. 7 (March 01, 2022) 752-761. Published online January 06, 2022.
KISQALI® Plus FEMARA® Improves Overall Survival in Advanced Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 290,560 new cases of breast cancer will be diagnosed in 2022 and about 43,780 individuals will die of the disease, largely due to metastatic recurrence. Approximately 70% of breast tumors express Estrogen Receptors and/or Progesterone Receptors and these patients are often treated with anti-estrogen therapy as first line treatment. However, resistance to hormonal therapy occurs in a majority of the patients.
Cyclin Dependent Kinases (CDK) play a very important role to facilitate orderly and controlled progression of the cell cycle. Genetic alterations in these kinases and their regulatory proteins have been implicated in various malignancies. Cyclin Dependent Kinases 4 and 6 (CDK4 and CDK6) phosphorylate RetinoBlastoma protein (RB), and initiate transition from the G1 phase to the S phase of the cell cycle. RetinoBlastoma protein has antiproliferative and tumor-suppressor activity and phosphorylation of RB protein nullifies its beneficial activities. CDK4 and CDK6 are activated in hormone receptor positive breast cancer, promoting breast cancer cell proliferation. Further, there is evidence to suggest that endocrine resistant breast cancer cell lines depend on CDK4 for cell proliferation and associated with increased expression of CDK4. The understanding of the role of Cyclin Dependent Kinases in the cell cycle, has paved the way for the development of CDK inhibitors.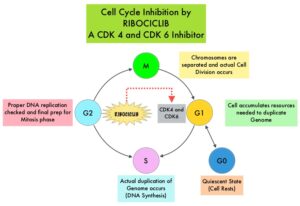
KISQALI® (Ribociclib) is an orally bioavailable, selective, small-molecule inhibitor of CDK4/6, preferentially inhibiting CDK4, that blocks the phosphorylation of RetinoBlastoma protein, thereby preventing cell-cycle progression and inducing G1 phase arrest. In a phase 1b study involving postmenopausal women with ER positive, HER2-negative advanced breast cancer, KISQALI® in combination with FEMARA® (Letrozole) demonstrated an Overall Response Rate (ORR) of 46% and a Clinical Benefit Rate of 79%, in treatment-naïve patients with advanced breast cancer. This led to the design of MONALEESA-2 trial.
MONALEESA-2 trial is a randomized, double-blind, placebo-controlled, Phase III study in which 668 patients were randomly assigned in a 1:1 ratio to receive either KISQALI® plus FEMARA® or placebo plus FEMARA®. Eligible patients included post-menopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer who had received no prior therapy for advanced disease. Treatment consisted of oral KISQALI® 600 mg daily on a 3-weeks on and 1-week off schedule, in 28-day treatment cycles plus FEMARA® 2.5 mg orally daily on a continuous schedule or placebo plus FEMARA®. Patients were stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression or unacceptable toxicity. No treatment crossover was allowed. The median age was 62 years, close to 60% of the patients had visceral metastases, and patients were stratified according to the presence or absence of liver or lung metastases. The Primary end point was Progression Free Survival (PFS) and Secondary end points included Overall Survival (OS), Overall Response Rate (ORR), Clinical Benefit Rate (Overall Response plus Stable disease lasting 24 weeks or more), Safety, and Quality of Life assessments.
In the primary and updated analyses of the MONALEESA-2 trial, PFS was significantly longer with KISQALI® plus FEMARA® than with placebo plus FEMARA® (25.3 months versus 16.0 months; HR for disease progression or death=0.57; P<0.001). The Overall Survival data were immature at the time of the primary and updated analyses. The authors have now reported the findings from the protocol-specified final analysis of Overall Survival, which is a key Secondary end point.
After a median follow up of 6.6 years, a significant Overall Survival benefit was observed with KISQALI® plus FEMARA®, compared to placebo plus FEMARA®. The median Overall Survival was 63.9 months with KISQALI® plus FEMARA® and 51.4 months with placebo plus FEMARA® (HR=0.76; two-sided P=0.008). This Overall Survival benefit was consistent across all prespecified subgroups. The median time to first subsequent chemotherapy was 50.6 months in the KISQALI® group and 38.9 months in the placebo group (HR for receipt of first chemotherapy=0.74). No new safety signals were observed.
It was concluded from the analysis of the MONALEESA-2 trial that first line therapy with KISQALI® plus FEMARA® showed a significant Overall Survival benefit as compared with placebo plus FEMARA®, in patients with HR-positive, HER2-negative advanced breast cancer, with a 24% relative reduction in the risk of death. The authors added that MONALEESA trials of KISQALI® have shown a consistent Overall Survival benefit regardless of accompanying endocrine therapy, line of therapy, or menopausal status.
Overall Survival with Ribociclib plus Letrozole in Advanced Breast Cancer. Hortobagyi GN, Stemmer SM, Burris HA, et al. N Engl J Med 2022; 386:942-950
FDA Approves LAG-3 Inhibitor OPDUALAG® and OPVIDO® in Advanced Untreated Melanoma
SUMMARY: The FDA on March 18, 2022, approved OPDIVO® (Nivolumab) and OPDUALAG® (Relatlimab-rmbw), for adult and pediatric patients 12 years of age or older, with unresectable or metastatic melanoma. The American Cancer Society’s estimates that for 2022, about 99,780 new cases of melanoma of the skin will be diagnosed in the United States and 7,650 people are expected to die of the disease. The rates of melanoma have been rising rapidly over the past few decades, but this has varied by age.
A better understanding of Immune checkpoints has opened the doors for the discovery of novel immune targets. Immune checkpoints are cell surface inhibitory proteins/receptors that harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies have been developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By blocking the Immune checkpoint proteins, T cells are unleashed, resulting in T cell proliferation, activation and a therapeutic response.
YERVOY® (Ipilimumab), a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4 was compared with PD-1 inhibitors, OPDIVO® (Nivolumab) and KEYTRUDA® (Pembrolizumab) in patients with advanced melanoma, and both OPDIVO® and KEYTRUDA® have demonstrated superior Overall Survival (OS), Progression Free Survival (PFS), and Objective Response Rate (ORR), and with a better safety profile. In the CheckMate 067, which is a double-blind Phase III study, results from the 6.5 year analysis showed that a combination of OPDIVO® plus YERVOY® demonstrated significant improvement in OS and PFS, when compared to single agent OPDIVO® or single agent YERVOY®.
In an attempt to improve outcomes and enhance the risk-benefit profiles of immunotherapy combinations, alternate Immune checkpoints are being explored. LAG-3 (Lymphocyte-Activation Gene 3 (LAG-3), is a cell-surface receptor expressed on immune cells including activated CD4+ T cells, and negatively regulates T-cell proliferation, inhibits T-cell activation and effector T-cell function. LAG-3 is upregulated in several tumor types, including malignant melanoma.
OPDUALAG® (Relatlimab) is a first-in-class human IgG4 LAG-3–blocking antibody that binds to LAG-3 and restores the effector function of exhausted T cells, resulting in T cell proliferation, activation and a therapeutic response. In preclinical studies, dual inhibition of LAG-3 and PD-1 showed synergistic antitumor activity, and in a Phase I/II trial, the combination of OPDUALAG® and OPDIVO®, demonstrated durable Objective Responses in patients with Relapsed/Refractory melanoma following treatment with PD-1 inhibitors.
RELATIVITY-047 is a Phase II/III, global, multicenter, double-blind, randomized trial in which a fixed-dose combination of OPDUALAG® and OPDIVO® was compared with OPDIVO® alone, in patients with previously untreated metastatic or unresectable melanoma. In this study, 714 patients were randomly assigned 1:1 to receive OPDUALAG® 160 mg and OPDIVO® 480 mg in a fixed-dose combination (N=355) or single agent OPDIVO® 480 mg (N=359). Both regimens were administered as an IV infusion over 60 minutes every 4 weeks, and treatment was continued until disease progression, unacceptable toxicities, or withdrawal of consent. Both treatment groups were well balanced and patients were stratified according to LAG-3 expression (1% or more versus less than 1%), PD-L1 expression (1% or more versus less than 1%), BRAF V600 mutation status, and metastasis stage (M0 or M1 with normal LDH levels versus M1 with elevated LDH levels). More patients in the OPDUALAG®- OPDIVO® group had Stage M1c disease, and a larger proportion had three or more sites with at least one metastatic lesion. The Primary end point was Progression Free Survival (PFS) as assessed by blinded Independent Central Review. Secondary end points included Overall Survival and Objective Response Rate (ORR). The median follow up was 13.2 months and the use of subsequent therapies upon progression was similar in the two treatment groups.
The median PFS was 10.1 months with OPDUALAG®- OPDIVO® as compared with 4.6 months with OPDIVO® (HR=0.75; P=0.006). The PFS benefit at 12 months with OPDUALAG®- OPDIVO® was 47.7% compared to 36.0% with OPDIVO®. The PFS benefit was more so with Relatlimab- OPDIVO® across key prespecified subgroups, compared to single agent OPDIVO®. Patients with poor prognosis characteristics, such as visceral metastases, high tumor burden, elevated levels of serum LDH, or mucosal or acral melanoma, had better outcomes with OPDUALAG®- OPDIVO® combination, than with single agent OPDIVO®. Further, a benefit with OPDUALAG®- OPDIVO® was also noted across BRAF mutant and wild-type subgroups, compared to OPDIVO®. Expression of LAG-3 or PD-L1 was not useful in predicting a benefit of OPDUALAG®- OPDIVO® over single agent OPDIVO® and appears to NOT have a clear role in treatment selection.
Grade 3 or 4 toxicities occurred in 18.9% of patients in the OPDUALAG®- OPDIVO® group and in 9.7% of patients in the single agent OPDIVO® group. The Safety profile of OPDUALAG®- OPDIVO® appeared favorable, when compared with dual checkpoint inhibition with a CTLA-4 inhibitor and PD-1 inhibitor combination (YERVOY®- OPDIVO®) in the CheckMate 067 trial, in which Adverse Events were noted in 59% of patients.
It was concluded that inhibition of two immune checkpoints, LAG-3 and PD-1, provided greater benefit with regards to Progression Free Survival, than inhibition of PD-1 alone, in patients with previously untreated metastatic or unresectable melanoma. The authors added that these results validate blocking LAG-3 in combination with PD-1 as a therapeutic strategy for patients with melanoma, and establishes LAG-3 as the third immune checkpoint pathway, thus providing more treatment options for patients with advanced melanoma.
Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. Tawbi HA, Schadendorf D, Lipson EJ, et al. for the RELATIVITY-047 Investigators. N Engl J Med 2022;386:24-34.
Efficacy and Safety in the Phase 3 QUAZAR® AML-001 Study of ONUREG® (azacitidine) Tablets

Written by: Thomas E Boyd, MD, Texas Oncology
Content Sponsored by: Bristol Myers Squibb
Dr. Boyd is a paid consultant for BMS and was compensated for his contribution in drafting this article.
Acute myeloid leukemia (AML) is an aggressive hematologic cancer with 5-year relative survival rates less than 30%.1 The journey for patients diagnosed with AML is very complex, and helping patients achieve remission is a major goal of initial treatment.2 Relapse after this initial therapy poses a significant roadblock to survival, as evidenced by high relapse rates and the current 5-year survival rate.1,3 There remains a critical need for therapies that extend survival.4
After a patient enters first remission, continued AML treatment is one avenue for appropriate patients.4 ONUREG® is the first and only FDA-approved continued AML treatment for adult patients with acute myeloid leukemia who achieved first complete remission (CR) or complete remission with incomplete blood count recovery (CRi) following intensive induction chemotherapy and are not able to complete intensive curative therapy.5
The efficacy and safety of ONUREG® was evaluated in the Phase 3 QUAZAR®AML-001 trial, where patients who completed intensive induction with or without consolidation therapy were randomized 1:1 to receive ONUREG or placebo orally on Days 1 through 14 of each 28-day cycle.5 Randomization was stratified by age at time of induction therapy, cytogenetic risk category at time of induction therapy, prior history of MDS/CMML, and receipt of consolidation therapy following induction therapy.5
Median OS, the primary endpoint in the QUAZAR®AML-001 trial, was over 2 years (24.7 months) in the ONUREG® arm and 14.8 months in the placebo arm (hazard ratio [HR] 0.69, 95% confidence interval [CI]: 0.55-0.86; P=0.0009; Figure 1).5 This statistically significant survival benefit of ~10 months for patients with AML in first remission who received ONUREG® compared to placebo demonstrated that QUAZAR® AML-001 trial met its primary endpoint.5 Survival estimates were 73% for ONUREG® (n=168; 95% CI: 67, 78) and 56% for placebo (n=127; 95% CI: 49, 62) at 1 year, and 51% for ONUREG® (n=115; 95% CI: 44, 57) and 37% for placebo (n=82; 95% CI: 31, 43) at 2 years.6 Analyses of survival estimates at 1 and 2 years were not designed to show a difference between treatment arms.
Additional analyses show the influence of ONUREG® across subgroups vs placebo for median OS (Figure 2). It is important to note that these prespecified analyses should be interpreted with caution because of potential selection bias, insufficient sample size, and a higher probability of making a false positive finding.7
The most common adverse reactions (ARs, ≥ 10%) associated with ONUREG® treatment included nausea, vomiting, diarrhea, fatigue/asthenia, constipation, pneumonia, abdominal pain, arthralgia, decreased appetite, febrile neutropenia, dizziness, and pain in extremity.5 Serious ARs occurred in 15% of patients who received ONUREG®, and the most common Grade 3/4 ARs were febrile neutropenia (11%), pneumonia (9%), diarrhea (5%), and fatigue/asthenia (4%).5
Efficacy and safety were established by the large, multicenter QUAZAR® AML-001 trial. These results established ONUREG® as a continued treatment for adult patients with AML who achieve first complete remission (CR) or complete remission with incomplete blood count recovery (CRi) following intensive induction chemotherapy and are not able to complete intensive curative therapy.
Figure 1: Kaplan-Meier curve for OS: ITT population in QUAZAR® AML-0015
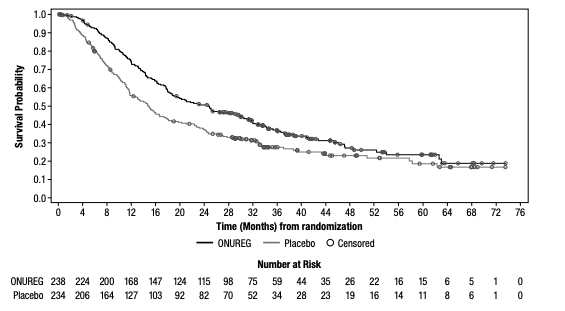
Figure 2: Overall survival select subgroup analysis6,7
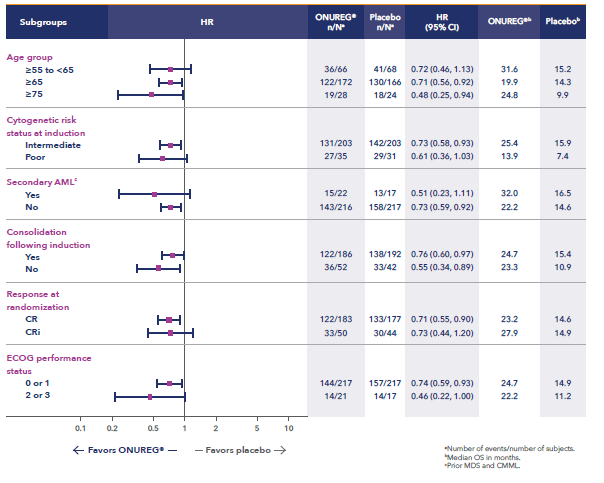
*Analysis limitations: These prespecified subgroup analyses should be interpreted with caution to determine a difference between arms in these select subgroups because of potential selection bias, insufficient sample size, and a higher probability of making a false positive finding.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
ONUREG® is contraindicated in patients with known severe hypersensitivity to azacitidine or its components.
WARNINGS AND PRECAUTIONS
Risks of Substitution with Other Azacitidine Products
Due to substantial differences in the pharmacokinetic parameters, the recommended dose and schedule for ONUREG® are different from those for the intravenous or subcutaneous azacitidine products. Treatment of patients using intravenous or subcutaneous azacitidine at the recommended dosage of ONUREG® may result in a fatal adverse reaction. Treatment with ONUREG® at the doses recommended for intravenous or subcutaneous azacitidine may not be effective. Do not substitute ONUREG® for intravenous or subcutaneous azacitidine.
Myelosuppression
New or worsening Grade 3 or 4 neutropenia and thrombocytopenia occurred in 49% and 22% of patients who received ONUREG®. Febrile neutropenia occurred in 12%. A dose reduction was required for 7% and 2% of patients due to neutropenia and thrombocytopenia. Less than 1% of patients discontinued ONUREG® due to either neutropenia or thrombocytopenia. Monitor complete blood counts and modify the dosage as recommended. Provide standard supportive care, including hematopoietic growth factors, if myelosuppression occurs.
Increased Early Mortality in Patients with Myelodysplastic Syndromes (MDS)
In AZA-MDS-003, 216 patients with red blood cell transfusion-dependent anemia and thrombocytopenia due to MDS were randomized to ONUREG® or placebo. 107 received a median of 5 cycles of ONUREG® 300 mg daily for 21 days of a 28-day cycle. Enrollment was discontinued early due to a higher incidence of early fatal and/or serious adverse reactions in the ONUREG® arm compared with placebo. The most frequent fatal adverse reaction was sepsis. Safety and effectiveness of ONUREG® for MDS have not been established. Treatment of MDS with ONUREG® is not recommended outside of controlled trials.
Embryo-Fetal Toxicity
ONUREG® can cause fetal harm when administered to a pregnant woman. Azacitidine caused fetal death and anomalies in pregnant rats via a single intraperitoneal dose less than the recommended human daily dose of oral azacitidine on a mg/m2 basis. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ONUREG® and for at least 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with ONUREG® and for at least 3 months after the last dose.
ADVERSE REACTIONS
Serious adverse reactions occurred in 15% of patients who received ONUREG®. Serious adverse reactions in ≥2% included pneumonia (8%) and febrile neutropenia (7%). One fatal adverse reaction (sepsis) occurred in a patient who received ONUREG®.
Most common (≥10%) adverse reactions with ONUREG® vs placebo were nausea (65%, 24%), vomiting (60%, 10%), diarrhea (50%, 21%), fatigue/asthenia (44%, 25%), constipation (39%, 24%), pneumonia (27%, 17%), abdominal pain (22%, 13%), arthralgia (14%, 10%), decreased appetite (13%, 6%), febrile neutropenia (12%, 8%), dizziness (11%, 9%), pain in extremity (11%, 5%).
LACTATION
There are no data regarding the presence of azacitidine in human milk or the effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with ONUREG® and for 1 week after the last dose.
Please see full Prescribing Information for ONUREG®.
References
1. National Cancer Institute. SEER Cancer Statistics Factsheets: Acute Myeloid Leukemia. http://seer.cancer.gov/statfacts/html/amyl.html. Accessed April 21, 2021.
2. Medeiros BC. Interpretation of clinical endpoints in trials of acute myeloid leukemia. Leuk Res. 2018;68:32-29.
3. Wei AH. Maintenance therapy for AML: are we there yet? Blood. 2019;133(13):1390-1392.
4. Medeiros BC, Chan SM, Daver NG, Jonas BA, Pollyea DA. Optimizing survival outcomes with post-remission therapy in acute myeloid leukemia. Am J Hematol. 2019;94:803-811.
5. ONUREG® [Prescribing Information]. Summit, NJ: Celgene Corporation; 2021.
6. Wei AH, Döhner H, Pocock C, et al. The QUAZAR AML-001 maintenance trial: results of a phase III international, randomized, double-blind, placebo-controlled study of CC-486 in patients with acute myeloid leukemia (AML) in first remission [oral presentation at ASH 2019]. Blood. 2019;134(Suppl 2):LBA-3.
7. Wei AH, Döhner H, Pocock C, et al. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N Engl J Med. 2020;383:2526-2537.
© 2021 Celgene Corporation.
ONUREG is a trademark of Celgene Corporation, a Bristol Myers Squibb company.
QUAZAR® is a registered trademark of Celgene Corporation, a Bristol Myers Squibb company
02/22 2011-US-2100200
Association of Gut Microbiome with Immune Checkpoint Inhibitor Response in Advanced Melanoma
SUMMARY: The American Cancer Society estimates that in 2022, there will be an estimated 1.92 million new cancer cases diagnosed and 609,360 cancer deaths in the United States. Immunotherapy with Immune Checkpoint Inhibitors (ICIs) has revolutionized cancer care and has become one of the most effective treatment options by improving Overall Response Rate and prolongation of survival across multiple tumor types. These agents target Programmed cell Death protein-1 (PD-1), Programmed cell Death Ligand-1 (PD-L1), Cytotoxic T-Lymphocyte-Associated protein-4 (CTLA-4), and many other important regulators of the immune system. Over 50% of patients treated with a combination of PD-1 and CTLA-4 inhibitors are alive after five years. Nonetheless, less than 50% of the patients respond to single-agent ICI and a higher response to targeting both PD-1 and CTLA-4 is associated with significant immune-related Adverse Events.
Biomarkers predicting responses to ICI’s include Tumor Mutational Burden (TMB), Mismatch Repair (MMR) status, and Programmed cell Death Ligand 1 (PD‐L1) expression. Other biomarkers such as Tumor Infiltrating Lymphocytes (TILs), TIL‐derived Interferon‐γ, Neutrophil‐to‐Lymphocyte ratio, and peripheral cytokines, have also been proposed as predictors of response. It has been postulated that concomitant medications during therapy with ICIs such as baseline steroid use as well as treatment with antibiotics may negate or lessen the efficacy of ICIs.
Preclinical studies have suggested that immune-based therapies for cancer may have a very complex interplay with the host’s microbiome and there may be a relationship between gut bacteria and immune response to cancer. The gut microbiome is unique in each individual, including identical twins. The crosstalk between microbiota in the gut and the immune system allows for the tolerance of commensal bacteria (normal microflora) and oral food antigens and at the same time enables the immune system to recognize and attack opportunistic bacteria. Immune Checkpoint Inhibitors strongly rely on the influence of the host’s microbiome, and the gut microbial diversity enhances mucosal immunity, dendritic cell function, and antigen presentation. Broad-spectrum antibiotics can potentially alter the bacterial composition and diversity of our gut microbiota, by killing the good bacteria. It has been postulated that this may negate the benefits of immunotherapy and influence treatment outcomes. It should be noted however that the relationship between gut bacteria and immune response is influenced by several factors and may be partially cancer type specific and it is unlikely that the same microbiome features can reflect the uniqueness of the genetic and immune characteristics of each tumor.
Even though the composition of the gut microbiome has been associated with clinical responses to immune checkpoint inhibitor (ICI) treatment, there is a lack of consistency of results between the published studies, and there is limited consensus on the specific microbiome characteristics linked to the clinical benefits of ICIs. The Predicting Response to Immunotherapy for Melanona with Gut Microbiome and Metabolomics (PRIMM) studies are two separate prospective observational cohort studies that has been recruiting patients in the UK (PRIMM-UK) and the Netherlands (PRIMM-NL) since 2018. These cohorts of previously ICI-naive patients with advanced melanoma have provided extensive biosamples, including stool, serum and peripheral blood mononuclear cells, before and during ICI treatment, with detailed clinical and dietary data collected at regular intervals longitudinally.
The authors therefore performed a meta-analysis on existing publicly available datasets to produce the largest study to date. In order to study the role of the gut microbiome in ICI response, the researchers recruited ICI-naive patients with advanced cutaneous melanoma from the PRIMM cohorts, as well as three additional cohorts of ICI-naive patients with advanced cutaneous melanoma, originating from Barcelona, Leeds and Manchester (N = 165), and performed shotgun metagenomic sequencing on a total of 165 stool microbiome samples collected before initiating ICI treatment. Shotgun sequencing is a laboratory technique for determining the DNA sequence of an organism’s genome. This dataset was integrated with 147 metagenomic samples from smaller publicly available datasets. This methodology provided the largest assessment of the potential of the gut microbiome as a biomarker of response to ICI, in addition to allowing for investigation of specific microbial species or functions associated with response. Patient demographics including age, gender, BMI, previous non-immunotherapy treatments, previous drug therapies such as antibiotics, Proton Pump Inhibitors (PPIs) and steroids, as well as dietary patterns, were collected in these cohorts for the majority of patients, and were considered in the multivariate analysis.
The researchers used machine learning analysis to understand the association between gut microbiome and response to ICIs. This analysis confirmed the link between the microbiome and Overall Response Rates (ORRs), as well as Progression Free Survival (PFS) with ICIs. This analysis also revealed limited reproducibility of microbiome-based signatures across cohorts. A panel of species, including Bifidobacterium pseudocatenulatum, Roseburia spp. and Akkermansiamuciniphila were associated with responders, but no single species could be regarded as a fully reliable biomarker across studies. Based on these findings from this large set of real-world cohorts, the authors noted that the relationship between human gut microbiome and response to ICIs is more complex than previously understood, and extends beyond the presence or absence of different microbial species in responders and nonresponders.
It was concluded that future studies should include large samples and take into account the complex interplay of clinical factors with the gut microbiome over the treatment course. Until then, the authors recommend high-quality, diverse, whole-foods diet to optimize gut health, rather than consumption of commercial probiotics.
Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Lee KA, Thomas AM, Bolte LA, et al. Nat Med. 2022 Feb 28. doi: 10.1038/s41591-022-01695-5. Online ahead of print.